
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben

Beginnen Sie mit dem Assemblierungsniveau, das Ihre Forschung tatsächlich benötigt.
Viele Seiten zur Genomassemblierung beginnen mit dem Vergleich von Sequenzierungsplattformen. In der Praxis ist der bessere Ausgangspunkt jedoch das Assemblierungsniveau. Ein mikrobielles Genom, ein erster Entwurf für eine Nicht-Modellart, ein Chromosomen-niveau Pflanzen-Genom und ein haplotyp-resolviertes Tiergenom benötigen nicht denselben Plan.
Bevor wir einen Arbeitsablauf empfehlen, helfen wir Ihnen, festzulegen, was die endgültige Assemblierung unterstützen muss. Ein Projekt zur Genentdeckung benötigt möglicherweise eine andere Assemblierungs- und Annotierungsstrategie als ein Projekt, das sich auf Merkmalskartierung, strukturelle Variation, Pan-Genom-Konstruktion oder Populationsgenomik konzentriert.
Entwurf oder Kontig-Ebene Assemblierung für frühe Genomressourcen
Ein Entwurf oder eine Contig-Level-Assemblierung kann geeignet sein, wenn Ihr Ziel eine frühe Referenzressource, umfassende Genentdeckung, Rekonstruktion mikrobieller Genome oder eine vorläufige vergleichende Analyse ist. Sie kann nützlich sein, wenn das Genom kompakt ist, die Forschungsfrage keine chromosomale Anordnung erfordert oder das Projekt als erster Schritt vor einer tiefergehenden Analyse konzipiert ist.
Dieses Niveau kann einen praktischen Ausgangspunkt bieten, unterstützt jedoch möglicherweise nicht vollständig die Verknüpfungsanalyse, die chromosomale strukturelle Interpretation oder die komplexe Wiederholungsauflösung.
Chromosomenebene-Assemblierung für Verknüpfungs-, Merkmals- und Vergleichsstudien
Eine Chromosomenebene-Assemblierung ist oft erforderlich, wenn die genomische Position von Bedeutung ist. Dazu gehören Merkmalskartierung, Züchtungsforschung, Chromosomenentwicklung, Syntenieanalyse, vergleichende Genomik und viele Pflanzen- oder Tiergenomprojekte.
Hi-C-Scaffolding kann helfen, assemblierte Contigs in Chromosomen-große Gerüste zu ordnen und auszurichten. Für Projekte, bei denen die endgültige Assemblierung die nachgelagerte Kartierung oder vergleichende Arbeiten unterstützen soll, kann eine Chromosomenstruktur wertvoller sein als eine fragmentierte Assemblierung mit hoher lokaler Genauigkeit, aber begrenzter langfristiger Organisation.
Haplotypen-aufgelöste Assemblierung für heterozygote oder polyploide Genome
Für hoch heterozygote, auskreuzende, hybride oder polyploide Organismen kann eine einzelne zusammengefasste Darstellung wichtige allele- oder haplotyp-spezifische Strukturen verbergen. In diesen Fällen kann eine haplotypaufgelöste oder phasierte Assemblierung nützlich sein.
Diese Strategie kann wichtig sein für die Pflanzen- und Tierzucht, die entwicklungsspezifische Genentdeckung, die Analyse struktureller Variationen und Projekte, bei denen Subgenome oder homologe Chromosomen sorgfältig interpretiert werden müssen.
T2T-ähnliche Assemblierung, wenn Wiederholungen, Zentromere und Telomere wichtig sind
Eine T2T-ähnliche Strategie kann in Betracht gezogen werden, wenn ungelöste Lücken, lange Wiederholungen, zentromerische Regionen, telomerische Regionen oder komplexe strukturelle Regionen im Mittelpunkt der Studie stehen. Dies ist in der Regel ein projektintensiverer Typ, da er stark von der Probenqualität, der Lese-Länge, der Assemblierungsstrategie und der manuellen oder maßgeschneiderten Überprüfung abhängt.
Nicht jedes Projekt benötigt eine T2T-ähnliche Assemblierung. Wir helfen Ihnen zu entscheiden, ob dieses Maß an Auflösung für Ihre Forschungsfrage notwendig ist oder ob eine Chromosomenebene oder eine phasierte Assemblierung praktischer wäre.
Ordnen Sie die Sequenzierungsstrategie der Genomkomplexität zu.
Sobald das Zielniveau der Assemblierung klar ist, wird die Sequenzierungsstrategie einfacher zu entwerfen. Verschiedene Genome erfordern unterschiedliche Evidenzschichten. Genomgröße, Wiederholungsinhalt, Ploidie, Heterozygotie, Kontaminationsrisiko und Probenqualität beeinflussen alle den endgültigen Assemblierungsplan.
Wenn PacBio HiFi der Genauigkeitsanker ist
PacBio SMRT-Sequenzierung kann Genome-Assemblierungsprojekte unterstützen, die lange Leseevidenz mit hoher Konsensgenauigkeit erfordern. PacBio HiFi-Lesungen sind oft wertvoll für die de novo Genome-Assemblierung, da sie lange Lese-Strukturen mit hoher Genauigkeit pro Leseeinheit kombinieren.
PacBio HiFi kann besonders nützlich sein, wenn das Projekt eine zuverlässige Konsensqualität, eine starke Rückgewinnung des Genraums und eine saubere Grundlage für die Annotation benötigt.
Wenn Nanopore-Ultra-Long-Reads helfen, Wiederholungen zu überbrücken
Nanoporen-Sequenzierung kann nützlich sein, wenn das Genom lange Wiederholungen, große strukturelle Regionen oder Lücken enthält, die längere Nachweise erfordern. Für einige komplexe Genome können ultralange Reads helfen, Regionen zu überbrücken, die kürzere Reads nicht auflösen können.
Nanopore-Daten können auch in T2T-ähnlichen Strategien oder Projekten berücksichtigt werden, bei denen die Leselänge ein großer Vorteil ist.
Wenn Hi-C für die Chromosomen-skalierte Strukturierung benötigt wird
Hi-C Sequenzierungsdienst bietet langfristige Kontaktinformationen, die helfen können, Contigs zu ordnen und in Chromosomen-große Gerüste zu integrieren. Dies ist besonders relevant, wenn die endgültige Zusammenstellung eine chromosomenebene Struktur benötigt.
Hi-C ist nicht einfach eine optionale Dekoration. Wenn das Forschungsziel von der Chromosomenorganisation im großen Maßstab abhängt, kann Hi-C oder ein anderer Ansatz zur langfristigen Strukturierung ein wesentlicher Bestandteil der Strategie sein.
Wann die Politur von Kurzlesungen oder hybride Beweise weiterhin hilfreich sind
Short-Read-Sequenzierung kann in Assemblierungsprojekten weiterhin von Wert sein. Sie kann je nach Projektgestaltung das Polieren, lokale Korrekturen, die Überprüfung von Kontaminationen, die Bewertung von Varianten oder ergänzende Analysen unterstützen.
Eine hybride Strategie kann nützlich sein, wenn ein Datentyp nicht jede Frage beantwortet. Es geht nicht darum, jede Technologie einzubeziehen, sondern die Evidenzschichten zu kombinieren, die zum Genom und zum nachgelagerten Ziel passen.
Was wir überprüfen, bevor wir einen Montageplan empfehlen
Ein guter Montageplan beginnt mit einer Risikobewertung. Wir möchten keinen hochwertigen Workflow empfehlen, den das Muster nicht unterstützen kann, noch einen minimalen Workflow, der die nachgelagerte Forschungsfrage nicht beantworten kann.
Arten, Genomgröße, Ploidie und Heterozygotie
Zunächst überprüfen wir den Organismus und alle verfügbaren Genominformationen. Nützliche Details umfassen die geschätzte Genomgröße, Ploidie, bekannten Wiederholungsgehalt, erwartete Heterozygotie, verwandte Referenzgenome und ob die Art domestiziert, wild, hybrid, inzuchtbedingt, auskreuzend oder polyploid ist.
Diese Details helfen zu bestimmen, ob das Projekt eine standardmäßige de-novo-Assemblierung, eine Chromosomenebene-Scaffolding, eine haplotypenaufgelöste Assemblierung oder eine fortgeschrittenere Strategie benötigt.
HMW-DNA-Qualität und Probenrisiko
Die Qualität von hochmolekularer DNA ist einer der wichtigsten Faktoren bei der Langzeitgenomassemblierung. Gewebetyp, Konservierungsmethode, Extraktionsschwierigkeit, DNA-Fragmentgröße, Verunreinigungen, Polysaccharide, Polyphenole, mikrobielle Kontamination und das Alter der Probe können alle die Bibliothekskonstruktion und die Kontinuität der Assemblierung beeinflussen.
Bei schwierigen Proben überprüfen wir die Machbarkeit, bevor wir die Montagestrategie festlegen.
Vorhandene Sequenzierungsdaten oder Entwurfsassemblierungen
Einige Projekte beginnen mit vorhandenen Daten. Möglicherweise haben Sie bereits kurze Reads, PacBio-Reads, Nanopore-Reads, Hi-C-Daten oder eine fragmentierte Entwurfsmontage.
In diesen Fällen können wir helfen zu bewerten, ob die Daten wiederverwendet, verbessert, strukturiert, verfeinert, annotiert oder in einen überarbeiteten Montageworkflow integriert werden können.
Nachgelagerte Ziele, die das Design der Montage beeinflussen
Die nachgelagerten Ziele sollten den Montageplan gestalten. Ein Genom, das für die Genannotation vorgesehen ist, könnte andere Prioritäten benötigen als eines, das für strukturelle Variationen, Pan-Genomanalysen, genomweite Assoziationen, Populationsgenomik oder die Entwicklung von Zuchtmarkern gedacht ist.
Wir überprüfen diese Ziele frühzeitig, damit die Assemblierung als nutzbare Genomressource gestaltet wird und nicht nur als FASTA-Datei.
Vergleich der Optionen für Strategien zur Genomassemblierung
Die beste Strategie zur Genomassemblierung hängt sowohl vom Genom als auch vom Forschungsziel ab. Die folgende Tabelle fasst gängige Optionen zusammen und zeigt, wie wir helfen, sie zu positionieren.
| Strategie | Bestmöglicher Anwendungsfall | Beispielanforderungsempfindlichkeit | Genomkomplexität anpassen | QC-Überlegungen | Bereitschaft im downstream-Bereich |
|---|---|---|---|---|---|
| Kurzlese-Entwurf-Zusammenstellung | Kompakte Genome, frühe Screening, einfache mikrobielle Projekte oder vorläufige Ressourcen | Moderat; kürzere DNA-Fragmente können je nach Projekt akzeptabel sein. | Begrenzt für hohe Wiederholungen, große Genome und komplexe Strukturen. | Benötigt Überprüfung der Abdeckung, Kontaminationsprüfung und Überprüfung der Vollständigkeit der Montage. | Kann grundlegende Genentdeckungen oder mikrobiologische Analysen unterstützen, ist jedoch bei komplexen nachgelagerten Strukturen eingeschränkt. |
| PacBio HiFi-Assemblierung | Genaues de novo-Assembly, Wiederherstellung des Genraums, Konstruktion von Referenzgenomen, phasierungsbereite Projekte | Benötigt geeignetes hochwertiges DNA. | Stark für viele Pflanzen-, Tier-, Pilz- und Nicht-Modellgenome | Bewerten Sie Contig N50, BUSCO, QV, Vollständigkeit, Kontamination und Phasierung, falls zutreffend. | Starke Grundlage für Annotation, vergleichende Genomik und viele Referenzgenomprojekte |
| Nanopore-Langzeit- oder Ultra-Langzeit-Assemblierung | Wiederholungsreiche Regionen, lange strukturelle Regionen, Lückenverschluss, T2T-ähnliche Strategien | Hochsensibel gegenüber der Qualität und Fragmentlänge von HMW-DNA | Stark, wenn die Länge der Lesespanne entscheidend ist | Bewerten Sie die Lese-länge, Abdeckung, Konsensqualität, Wiederholungsauflösung und Polierstrategie. | Nützlich für komplexe Strukturen, Lückenauflösung und die Architektur des Genoms über lange Strecken. |
| Hi-C Gerüstbau | Chromosomenebene Assemblierung, Verknüpfung, Syntenie, Züchtung, vergleichende Genomik | Benötigt geeignetes Material für die Hi-C-Bibliotheksvorbereitung. | Stark für die Anordnung und Orientierung von Contigs in chromosomengroße Gerüste. | Bewerten Sie die Qualität der Kontaktkarte, die Genauigkeit des Gerüsts, Fehlzuordnungen und die Chromosomenzuweisung. | Wichtig für nachgelagerte Arbeiten auf Chromosomenebene |
| Hybride Montage | Projekte, die ergänzende Genauigkeit, Kontinuität, Verfeinerung oder langfristige Beweise benötigen | Hängt von allen enthaltenen Datentypen ab. | Flexibel für komplexe oder hochpreisige Projekte | Erfordert sorgfältige Integration und plattformübergreifende Qualitätskontrolle. | Stark, wenn die Versammlung mehrere nachgelagerte Anwendungen unterstützen muss. |
| Haplotypen-resolvierter Zusammenbau | Heterozygote, Hybrid, Mischlings- oder Polyploid-Organismen | Erfordert starke Datenqualität und ausreichende Abdeckung. | Stark, wenn allele-spezifische oder subgenom-sensible Interpretationen wichtig sind | Bewerten Sie die Phasengenauigkeit, Haplotyptrennung, Duplikation und Vollständigkeit. | Nützlich für Zucht, allelspezifische Analyse, SV und komplexe Genominterpretation. |
| T2T-ähnliche Montage | Zentromere, Telomere, lange Wiederholungen, ungelöste Lücken, hochwertige Referenzressourcen | Sehr empfindlich gegenüber der Probenqualität, der Leselänge und dem Datenlayout. | Stark für schwierige, sich wiederholende Bereiche, wenn sie durch Daten unterstützt werden. | Bewerten Sie die Schließung von Lücken, die Wiederholungsauflösung, die QV, die manuelle Überprüfung und die strukturelle Konsistenz. | Nützlich für hochwertige Referenzprojekte und wiederholungszentrierte Forschung |
| Mikrobielle, pilzliche oder kompakte Genomassemblierung | Bakterielle, pilzliche, virale, Plasmid- oder gentechnisch veränderte Stammgenome | Oft weniger anspruchsvoll als große eukaryotische Genome, aber die Kontrolle von Kontaminationen ist wichtig. | Geeignet für kompakte Genome; die Strategie hängt von Plasmiden, Wiederholungen und der Genomstruktur ab. | Bewerten Sie die Zirkularisierung, Kontamination, Plasmide, Vollständigkeit und die Qualität der Annotation. | Nützlich für die Stammcharakterisierung, vergleichende Genomik und Forschung in der synthetischen Biologie. |
End-to-End-Workflow von der Probenprüfung bis zur nutzbaren Genomressource
Von der Machbarkeitsprüfung über das Sequenzierungsdesign, die Assemblierung, die Qualitätskontrolle, die Annotation bis hin zu downstream-bereiten Dateien.
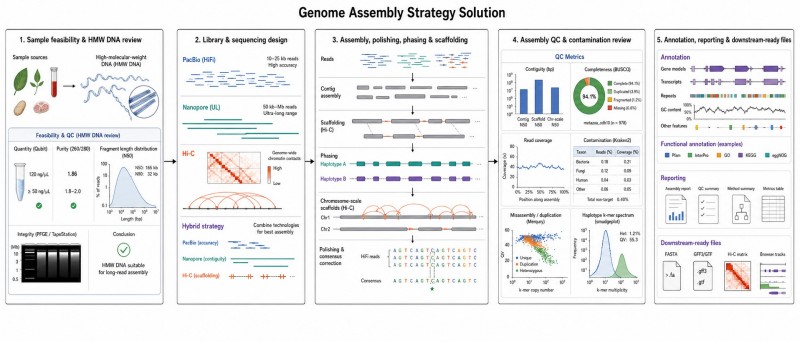
Ein Genomassemblierungsprojekt durchläuft mehrere technische und Entscheidungsprüfungen. Wir gestalten den Workflow um die endgültige Assemblierungsstufe und das nachgelagerte Forschungsziel.
Wir überprüfen den Organismus, den Proben-Typ, die Konservierungsmethode, die erwartete DNA-Qualität und Risikofaktoren. Für die Langlese-Assemblierung ist oft DNA mit hoher Molekulargewicht entscheidend. Wenn das Risiko der Probe hoch ist, besprechen wir die Optionen, bevor die Sequenzierung beginnt.
Basierend auf dem Zielversammlungsniveau empfehlen wir die benötigten Datentypen. Dazu können PacBio HiFi, Oxford Nanopore, Hi-C, Kurzlesepolitur oder ein hybrides Design gehören. Der Sequenzierungsplan sollte der Genomkomplexität entsprechen und nicht einem festen Template folgen.
Der Zusammenstellungsworkflow kann die Contig-Zusammenstellung, Politur, Haplotyp-Trennung, Scaffold-Konstruktion, Hi-C-basierte Anordnung und Orientierung, Lückenüberprüfung und T2T-ähnliche Verfeinerung umfassen, wenn dies angemessen ist.
Bei der Einbeziehung in das Projekt unterstützen wir wiederholte Annotationen, Genvorhersagen, funktionale Annotationen und nachgelagerte Bioinformatik. Die endgültige Ausgabe kann Assemblierungsdateien, Annotierungsdateien, QC-Berichte, visuelle Zusammenfassungen und Projektdokumentationen umfassen.
Musteranforderungen und Projektaufnahmeinformationen
Die Qualität der Proben hat einen direkten Einfluss auf die Strategie zur Genomassemblierung. Langleseassemblierung, scaffolding auf Chromosomenebene, haplotypbewusste Projekte und T2T-ähnliche Arbeitsabläufe können unterschiedliche Proben- und Datenplanungen erfordern.
Die endgültigen Anforderungen hängen von der Art, der Genomgröße, der Ploidie, dem Gewebetyp, der Konservierungsmethode, der Plattformwahl und dem angestrebten Zusammenstellungsniveau ab. Vor der Projektbestätigung überprüft unser Team die folgenden Informationen und empfiehlt den am besten geeigneten Arbeitsablauf.
| Muster- oder Eingabetyp | Was wir überprüfen | Qualitätsfokus | Erforderliche Projektinformationen | Typische QC-Prüfpunkte | Notizen |
|---|---|---|---|---|---|
| Frisches oder gefrorenes Gewebe für HMW-DNA | Gewebetyp, Erhaltung, erwarteter DNA-Ertrag, Kontaminationsrisiko | Lange DNA-Fragmente geeignet für Langlesebibliotheken | Art, Schätzung der Genomgröße, Ploidie, nachgelagertes Ziel | DNA-Integrität, Reinheit, Konzentration, Fragmentgröße, Kontaminationsüberprüfung | Die endgültigen Anforderungen hängen von der Art, der Genomgröße, dem Assemblierungsniveau und der Plattformstrategie ab. |
| Pflanzen-, Tier-, Pilz- oder Nicht-Modellorganismenproben | Beispielquelle, Gewebeschwierigkeit, Inhibitoren, verwandte Referenzen, erwarteter Wiederholungsinhalt | Machbarkeit für de novo, chromosomenebene oder phasierte Assemblierung | Art, Probenquelle, geschätzte Genomgröße, Ploidie, Ziel-Assemblierungsniveau | Stichproben-Eignungsprüfung, DNA-Qualitätsprüfung, Kontaminationsrisikoprüfung | Komplexe oder inhibitorreiche Gewebe erfordern möglicherweise eine spezielle Überprüfung vor der Auswahl des Workflows. |
| Vorhandene Sequenzierungsdaten | FASTQ/BAM-Dateien, Plattform, Abdeckung, Lese-Länge, Probenbezeichnungen, vorherige Assemblierung | Kompatibilität mit Wiederzusammenbau, Polieren, Gerüstbau oder Annotation | Sequenzierungsplattform, Genomziel, vorherige Assemblierungsdateien, Analyseziel | Dateiintegrität, Lese-QC, Abdeckungsüberprüfung, Machbarkeitsprüfung der Montage | Kann Unterstützung bei Rettung, Verbesserung, Neu-Analyse oder nachgelagerter Annotation bieten, wenn die Datenqualität geeignet ist. |
| Entwurf von Assemblierungsdateien | Assembly FASTA, Statistiken, Annotationsstatus, Kontaminationsbedenken, Scaffold-Bedarf | Verbesserungspotenzial und Eignung für nachgelagerte Anwendungen | Assembly-FASTA, vorhandene QC, Arteninformation, gewünschtes Verbesserungsniveau | Kontinuitätsüberprüfung, BUSCO-Überprüfung, Kontaminationsprüfung, Machbarkeitsprüfung von Gerüsten | Kann je nach Daten durch Polieren, Gerüstbau, Annotation oder benutzerdefinierte Bioinformatik verbessert werden. |
Wie man die Qualitätskontrolle von Genomassemblierungen liest, ohne sich zu sehr auf N50 zu verlassen.
N50 wird häufig verwendet, sollte jedoch nicht die einzige Kennzahl sein, um eine Genomassemblierung zu bewerten. Ein hoher N50 kann lange Contigs oder Scaffolds widerspiegeln, bedeutet jedoch nicht automatisch, dass die Assemblierung vollständig, genau, korrekt strukturiert oder für jede nachgelagerte Analyse nützlich ist.
| QC-Metrik | Was es hilft zu bewerten | Was es nicht vollständig beantwortet. |
|---|---|---|
| Contig N50 | Montagekontinuität vor dem Gerüstbau | Vollständigkeit, Richtigkeit, Kontamination oder Genrückgewinnung |
| Gerüst N50 | Langstrecken-Gerüstkontinuität | Ob die Gerüste korrekt bestellt und ausgerichtet sind |
| SUCHEN | Gene-Raum-Vollständigkeit unter Verwendung konservierter Gene | Wiederholungsauflösung, strukturelle Korrektheit oder Ganzgenomgenauigkeit |
| QV | Konsensusschätzgenauigkeit | Langstreckenstruktur, Phasierungsqualität oder Nützlichkeit der Annotation |
| Vergleich der Genomgröße | Ob die Größe der Baugruppe den Erwartungen entspricht | Ob die Sequenz vollständig oder korrekt zusammengesetzt ist |
| Kontaminationsüberprüfung | Nicht-Zielsequenz- oder Mischprobenrisiko | Biologische Interpretation oder Annotierungsgenauigkeit allein |
| Hi-C Kontaktkartenübersicht | Konsistenz der Chromosomenebene-Scaffolding | Basisgenauigkeit oder Genvollständigkeit |
| Annotationszusammenfassung | Bereitschaft zur Genvorhersage und funktionalen Interpretation | Ob die Struktur der Baugruppe vollständig korrekt ist |
N50 kann helfen, die Kontinuität der Assemblierung zu beschreiben, misst jedoch nicht alles. Eine Assemblierung mit hohem N50 kann dennoch Kontamination, Fehlverbindungen, fehlende Gene, kollabierte Wiederholungen oder eine unzureichende Annotationsbereitschaft aufweisen.
BUSCO hilft dabei, die Vollständigkeit konservierter Gene zu bewerten, während QV eine Schätzungen zur Konsensgenauigkeit liefern kann, wenn dies zutrifft. Diese Metriken ergänzen N50, insbesondere wenn die Assemblierung die Entdeckung von Genen, vergleichende Genomik oder forschungsorientierte Veröffentlichungen unterstützen soll.
Der beste QC-Rahmen hängt davon ab, was die Assemblierung unterstützen muss. Ein Genom, das für die Genannotierung, Pan-Genom-Analyse, strukturelle Variation oder Merkmalskartierung verwendet wird, benötigt möglicherweise unterschiedliche Prüfungen. Wir helfen dabei, QC im Kontext des Forschungsziels zu interpretieren.
Annotation und nachgelagerte Analyse machen die Assemblierung nutzbar.
Eine Genomassemblierung wird wertvoller, wenn sie mit Annotation und nachgelagerten Analysen verbunden ist. Für viele Forschungsteams ist das endgültige Ziel nicht nur eine FASTA-Datei. Es ist eine nutzbare Genomressource.
Genomannotation und Genvorhersage
Wir können unterstützen. Genomannotation und Genvorhersagedienst für Projekte, die Genmodelle, kodierende Sequenzen, Proteinsequenzen, funktionale Annotationen und Zusammenfassungen der Annotationen erfordern.
Dies ist besonders wichtig für Nicht-Modellorganismen, Arten mit begrenzten Annotierungsressourcen und Projekte, die sich auf die Genentdeckung konzentrieren.
Wiederholungsannotation und funktionale Annotation
Die Wiederholungsannotation hilft dabei, transponierbare Elemente, sich wiederholende Regionen und Wiederholungsinhalte zu charakterisieren, die die Zusammenstellungsstrategie und die nachfolgende Interpretation beeinflussen können. Die funktionale Annotation kann dabei helfen, vorhergesagte Gene mit bekannten Datenbanken, Wegen, Genfamilien oder biologischen Funktionen zu verbinden.
Vergleichende Genomik, Pan-Genom, SV und Populationsunterstützung
Wenn die Versammlung nachgelagerte Studien unterstützen wird, können wir bei der Planung zusätzlicher Analysen helfen durch Genomdatenanalyse, Pan-Genom, Variantenerkennungund Populationsgenetik Dienstleistungen.
Diese Module können vergleichende Genomik, Erweiterung von Genfamilien, Konstruktion von Pan-Genomen, Analyse struktureller Variationen, Populationsgenomik und zuchtbezogene Forschung unterstützen.
Dateien, die Ihr Team für zukünftige Studien wiederverwenden kann
- Assembly FASTA
- GFF- oder GTF-Annotationsdateien
- Wiederholte Annotationsdateien
- Protein-FASTA und CDS-FASTA
- BUSCO-Berichte und QV-Zusammenfassungen
- Hi-C Gerüstausgaben
- Vergleichende Genomik-Tabellen
- Pan-Genom oder SV-bereite Dateien
- Projektbericht
Wählen Sie eine Strategie basierend auf der Forschungsfrage, nicht auf dem Technologietitel.
Eine gute Assemblierungsstrategie beginnt mit der Forschungsfrage. Wir helfen Ihnen zu entscheiden, welche Genomressource benötigt wird und welche Datentypen sie unterstützen können.
Wenn Ihr Ziel ein erstes Referenzgenom ist
Eine de novo Referenzstrategie kann geeignet sein, wenn kein naher Referenzgenom vorhanden ist oder wenn Sie eine neue Genomressource für eine Nicht-Modellart benötigen. In vielen Fällen, De Novo Whole Genome Sequenzierungsdienst oder Pflanzen-/Tier-Ganzgenom de novo Sequenzierung kann dieses Ziel unterstützen.
Wenn Ihr Ziel die Merkmalszuordnung oder Zuchtunterstützung ist
Die Chromosomenebene der Assemblierung kann nützlicher sein, wenn die genomische Position von Bedeutung ist. Hi-C-Scaffolding kann die Trait-Kartierung, die Verknüpfungsanalyse, die vergleichende Genomik und die züchtungsbezogene Forschung unterstützen.
Wenn Ihr Ziel eine polyploide oder haplotypbewusste Interpretation ist
Haplotyp-resolute Assemblierung kann erforderlich sein, wenn der Organismus hoch heterozygot, aus einer Kreuzung, hybrid oder polyploid ist. Diese Strategie kann helfen, allele- oder subgenomspezifische Strukturen zu bewahren, wenn sie durch Daten unterstützt wird.
Wenn Ihr Ziel das Pan-Genom, SV oder Populationsgenomik ist
Wenn die Assemblierung den Bau eines Pan-Genoms, die Analyse struktureller Variationen oder die Populationsgenomik unterstützen soll, helfen wir dabei, die Assemblierung und die nachfolgenden Ergebnisse gemeinsam zu planen. Das Ziel ist es, eine Assemblierung zu vermeiden, die auf dem Papier akzeptabel aussieht, aber für den nächsten Analyse-Schritt nicht geeignet ist.
Referenzen
- Benchmarking von Hi-C-Tools zur Strukturierung von Pflanzengenomen, die aus PacBio HiFi- und ONT-Reads gewonnen wurden.
- Telomer-zu-Telomer-Assemblierung diploider Chromosomen mit Verkko
- Skalierbare Telomer-zu-Telomer-Assemblierung für diploide und polyploide Genome mit doppeltem Graphen
- Chromosomen-große Assemblierungskomparaison des koreanischen Referenzgenoms KOREF von PromethION und PacBio mit Hi-C-Kartierungsinformationen
- Benchmarking von Hi-C-Tools zur Strukturierung von Pflanzengenomen, die aus PacBio HiFi- und ONT-Reads gewonnen wurden — PMC-Datensatz
Einhaltung / Haftungsausschluss
CD Genomics bietet diesen Service nur für Forschungszwecke (RUO) an. Dieser Service ist nicht für klinische Diagnosen, direkte medizinische Interpretationen, Patientenmanagement, Behandlungsanleitungen, Tests für Endverbraucher oder garantierte Entdeckungsansprüche gedacht.
Demonstrationsergebnisse
Demonstrationsergebnisse helfen Ihrem Team zu verstehen, was ein Montageprojekt liefern kann. Diese Beispiele zeigen Ausgabetypen, nicht feste biologische Schlussfolgerungen.
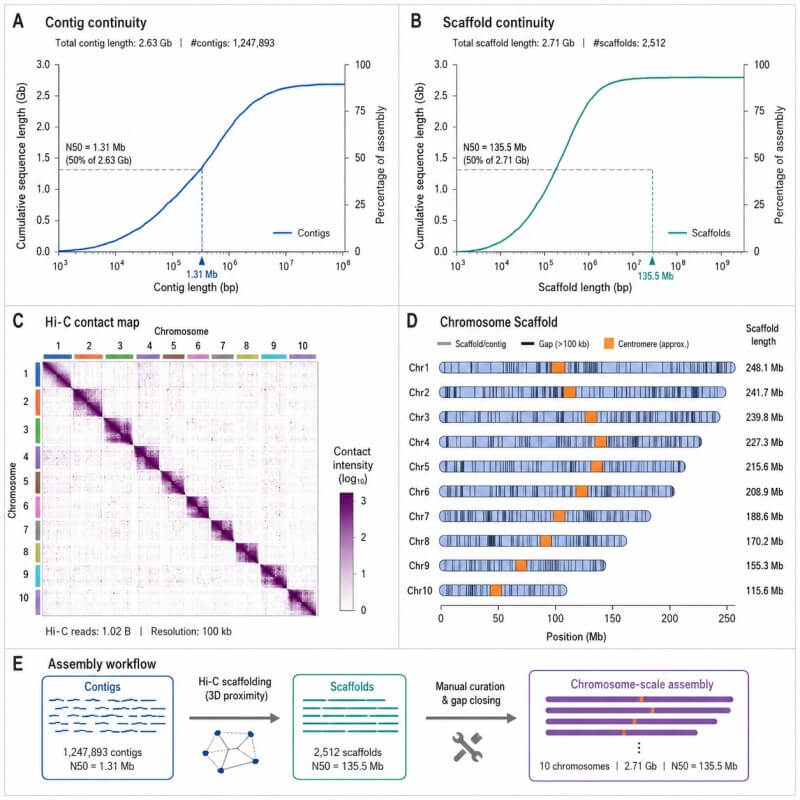
Zusammenfassung der Assemblierungs-Kontinuität und Chromosomen-Gerüstbildung
Diese Ausgabe kann Contig-Statistiken, Scaffold-Statistiken, Scaffold-Ansichten auf Chromosomenebene und eine Zusammenfassung der Hi-C-Kontaktkarte anzeigen, wenn Hi-C-Scaffolding enthalten ist.
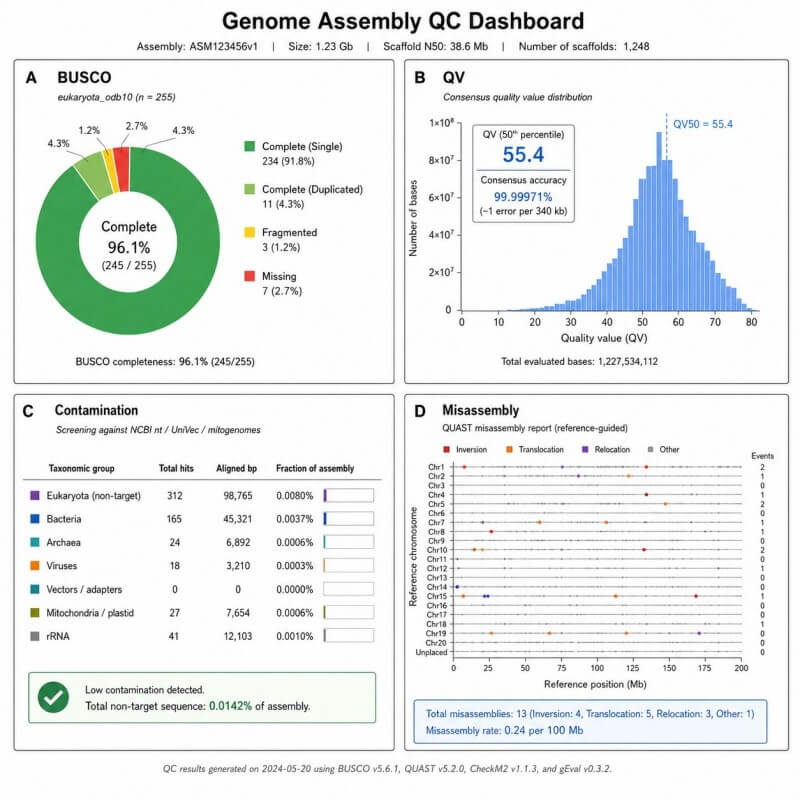
BUSCO, QV und Kontaminationsüberprüfungs-Dashboard
Dieser Bericht fasst die Vollständigkeit der Assemblierung, die Qualität des Konsenses und die Überprüfung der Kontamination in einem kompakten Format zusammen.

Annotation und downstream-bereite Ausgabesicht
Diese Ausgabe kann Zusammenfassungen der Genannotation, Wiederholungsannotierungsspuren, Ausgaben von Genfamilien und Dateien enthalten, die für vergleichende oder populationsbezogene Analysen vorbereitet wurden.
Häufig gestellte Fragen
1. Was ist eine Genom-Assemblierungsstrategie-Lösung?
Es handelt sich um einen forschungsorientierten Serviceansatz, der Ihnen hilft, den richtigen Genom-Assemblierungsplan basierend auf Ihrer Art, der Probenqualität, der Genomkomplexität, dem Assemblierungsniveau und den nachgelagerten Zielen auszuwählen und umzusetzen.
2. Wie finde ich heraus, welches Assemblierungsniveau mein Projekt benötigt?
Das richtige Niveau hängt von der Forschungsfrage ab. Die frühe Genentdeckung benötigt möglicherweise eine Entwurf- oder Contig-Assemblierung, während die Merkmalskartierung, Syntenie und Züchtungsforschung oft von einer Chromosomen-Assemblierung profitieren. Haplotype-resolvierte oder T2T-ähnliche Strategien können für komplexe Genome oder repeat-reiche Regionen in Betracht gezogen werden.
3. Wann ist die Entwurfszusammenstellung ausreichend?
Der Entwurf einer Assemblierung kann für kompakte Genome, die frühe Referenzentwicklung, vorläufige Genentdeckung oder Projekte, bei denen die chromosomale Position nicht zentral ist, ausreichend sein. Für Verknüpfungen, strukturelle Variationen, Chromosomenentwicklung oder Pan-Genom-Arbeiten könnte es jedoch nicht ausreichen.
4. Wann sollte ich eine Chromosomenebene-Assemblierung wählen?
Die Chromosomenebene der Assemblierung ist nützlich, wenn die genomische Position, die langfristige Struktur, die Merkmalszuordnung, die Syntenie oder die vergleichende Genomik von Bedeutung sind. Hi-C oder verwandte Scaffold-Methoden können verwendet werden, um diese Ebene zu unterstützen.
5. Wann ist eine haplotypenauflösende Assemblierung wichtig?
Haplotype-resolute Assemblierung kann wichtig für heterozygote, ausgemischte, hybride oder polyploide Organismen sein. Sie hilft, allele- oder haplotyp-spezifische Informationen zu bewahren, wenn die Daten und das Projektdesign dies unterstützen.
6. Wann ist die T2T-ähnliche Assemblierung in Betracht zu ziehen?
Eine T2T-ähnliche Strategie könnte in Betracht gezogen werden, wenn Zentromere, Telomere, große Wiederholungen, ungelöste Lücken oder eine hohe Qualität des Referenzgenoms zentral für die Forschungsfrage sind. Sie ist anspruchsvoller und sollte sorgfältig geplant werden.
7. Wie unterscheiden sich PacBio und Nanopore bei der Genomassemblierung?
PacBio HiFi-Lesungen werden oft wegen ihrer hohen Genauigkeit bei der Langleseanalyse geschätzt. Nanopore-Lang- oder Ultra-Lesungen können nützlich sein, um lange Wiederholungen und komplexe Regionen abzudecken. Viele Projekte profitieren davon, eine Technologie auszuwählen oder Technologien basierend auf dem Genom und dem Forschungsziel zu kombinieren.
8. Warum ist Hi-C nützlich für die Chromosomenebene Assemblierung?
Hi-C liefert Informationen über Langstrecken-Kontakte, die helfen können, Contigs in Chromosomen-große Gerüste zu ordnen und auszurichten. Es ist besonders nützlich, wenn die nachgelagerte Analyse von der Struktur auf Chromosomenebene abhängt.
9. Warum sollte ich mich nicht nur auf N50 verlassen?
N50 beschreibt die Kontinuität, misst jedoch nicht vollständig die Vollständigkeit, Genauigkeit, Kontamination, das Risiko von Fehlassemblierungen oder die Bereitschaft zur Annotation. Eine gründliche Qualitätskontrolle sollte mehrere Metriken kombinieren.
10. Welche Stichprobeninformationen werden benötigt, bevor eine Strategie empfohlen wird?
Nützliche Informationen umfassen Arten, Schätzung der Genomgröße, Ploidie, Probenart, Erhaltungsmethode, DNA-Qualität, erwartete Heterozygotie, verwandte Referenzgenome, vorhandene Sequenzierungsdaten und nachgelagerte Forschungsziele.
11. Können vorhandene Sequenzierungsdaten oder Entwurfassemblierungen verbessert werden?
Ja. Vorhandene Daten oder Entwurfsassemblierungen können das Polieren, Gerüstartikulation, die Wiederzusammenstellung, Annotation, Kontaminationsüberprüfung oder nachgelagerte Analysen unterstützen, wenn die Datenqualität geeignet ist.
12. Welche Ergebnisse kann ich von einem Genomassemblierungsprojekt erwarten?
Die Liefergegenstände können unter anderem assemblierte FASTA-Dateien, QC-Zusammenfassungen, N50-Statistiken, BUSCO-Berichte, QV-Schätzungen, Kontaminationsüberprüfungen, Annotationsdateien, Wiederholungsannotationen, Hi-C-Scaffolding-Ausgaben, für Genombrowser geeignete Dateien und Projektberichte umfassen.
13. Können Ergebnisse der Genomassemblierung Annotationen, Pan-Genome, SV oder Populationsgenomik unterstützen?
Ja. Wenn die Genomassemblierung richtig geplant wird, kann sie Annotation, vergleichende Genomik, Pan-Genom-Analyse, Analyse struktureller Variationen und Populationsgenomik unterstützen. Diese nachgelagerten Bedürfnisse sollten berücksichtigt werden, bevor der Assemblierungsplan finalisiert wird.
14. Ist dieser Dienst für klinische oder diagnostische Zwecke gedacht?
Nein. Dieser Dienst ist ausschließlich für forschungsorientierte Genomassemblierung und bioinformatische Projekte vorgesehen.
Literaturfall: Hi-C-Scaffolding verändert, wie Genomassemblierungen bewertet werden.
Veröffentlichte Forschungsübersicht
Tagebuch: Grenzen der Bioinformatik
Veröffentlicht: 2024
Hintergrund
Die Chromosomen-niveau Genomassemblierung erfordert oft mehr als nur die Erstellung von Contigs. Hi-C-Lesungen können helfen, große genomische Regionen in Gerüste zu ordnen und auszurichten, was sie nützlich für Projekte macht, die eine chromosomale Struktur benötigen.
Methoden
Die Studie erzeugte zwei de novo Assemblierungen von Arabidopsis thaliana aus denselben PacBio HiFi- und Oxford Nanopore-Daten. Anschließend wurden die Assemblierungen mit 3D-DNA, SALSA2 und YaHS strukturiert.
Die gerüsteten Assemblierungen wurden anhand von Kontinuität, Vollständigkeit, Genauigkeit und struktureller Korrektheit bewertet. Dieses Design ist relevant, da es nicht nur Sequenzierungsdatentypen vergleicht, sondern auch nachgelagerte Gerüstbildung und Qualitätsinterpretation.
Ergebnisse
- Die Studie berichtete, dass Hi-C-Scaffolding-Tools unterschiedliche Leistungsmerkmale in den bewerteten Assemblierungen zeigten.
- YaHS hat in dieser Analyse am besten abgeschnitten.
- Die umfassendere Lektion ist wichtig für die Projektplanung: Die Qualität der Chromosomenebene hängt nicht nur von der Sequenzierungsplattform ab, sondern auch von der Scaffold-Methode, der QC-Überprüfung und der strukturellen Korrektheit.
 Ein Hi-C-Scaffolding-Benchmark veranschaulicht, warum die Chromosomenebene der Genomassemblierung anhand von Kontinuität, Vollständigkeit, Genauigkeit und struktureller Korrektheit bewertet werden sollte, anstatt nur eine einzelne Kennzahl zu verwenden.
Ein Hi-C-Scaffolding-Benchmark veranschaulicht, warum die Chromosomenebene der Genomassemblierung anhand von Kontinuität, Vollständigkeit, Genauigkeit und struktureller Korrektheit bewertet werden sollte, anstatt nur eine einzelne Kennzahl zu verwenden.
Fazit
Dieser Fall unterstützt die zentrale Idee hinter unserer Lösung zur Genomassemblierung. Die Planung der Genomassemblierung sollte nicht bei der Auswahl von PacBio, Nanopore oder Hi-C enden. Eine starke Strategie berücksichtigt auch das Assemblierungsniveau, die Scaffolding-Methode, QC-Metriken, Annotation und die spätere Nutzbarkeit.


