

Abstract
Purpose:
Radiation-mediated immune suppression limits efficacy and is a barrier in cancer therapy. Radiation induces negative regulators of tumor immunity including regulatory T cells (Treg). Mechanisms underlying Treg infiltration after radiotherapy (RT) are poorly defined. Given that dendritic cells (cDC) maintain Treg we sought to identify and target cDC signaling to block Treg infiltration after radiation.
Experimental Design:
Transcriptomics and high dimensional flow cytometry revealed changes in murine tumor cDC that not only mediate Treg infiltration after RT, but associate with worse survival in human cancer datasets. Antibodies perturbing a cDC-CCL22-Treg axis were tested in syngeneic murine tumors. A prototype interferon-anti-epidermal growth factor receptor fusion protein (αEGFR-IFNα) was examined to block Treg infiltration and promote a CD8+ T cell response after RT.
Results:
Radiation expands a population of mature cDC1 enriched in immunoregulatory markers that mediates Treg infiltration via the Treg-recruiting chemokine CCL22. Blocking CCL22 or Treg depletion both enhanced RT efficacy. αEGFR-IFNα blocked cDC1 CCL22 production while simultaneously inducing an antitumor CD8+ T cell response to enhance RT efficacy in multiple EGFR-expressing murine tumor models, including following systemic administration.
Conclusions:
We identify a previously unappreciated cDC mechanism mediating Treg tumor infiltration after RT. Our findings suggest blocking the cDC1-CCL22-Treg axis augments RT efficacy. αEGFR-IFNα added to RT provided robust antitumor responses better than systemic free interferon administration, and may overcome clinical limitations to interferon therapy. Our findings highlight the complex behavior of cDC after RT and provide novel therapeutic strategies for overcoming RT-driven immunosuppression to improve RT efficacy.
INTRODUCTION
Radiotherapy (RT) has been used in the majority of cancer treatments for more than a century. RT reduces tumor burden and stimulates an antitumor immune response. (1–3) Preclinical evidence has revealed RT upregulates MHC class I presentation of tumor antigen, activates immunogenic cell death, and induces production of proinflammatory cytokines culminating in a CD8+ effector T cell response. (4, 5) Pharmacologic strategies to augment RT have focused on enhancing dendritic cell antigen presentation, subsequent CD8+ T cell cross-priming, and checkpoint blockade. (6–8) Though these approaches sparked initial excitement, trials combining concurrent RT and immunotherapy have had mixed success. (9–11) A growing body of evidence suggests that RT can simultaneously drive negative regulators of tumor immunity, including upregulation of immune checkpoints, recruitment of regulatory T cells (Treg), and infiltration of myeloid-derived suppressor cells (MDSC). (3, 12–16)
Radiation-driven Treg tumor accumulation and their suppressive role tumor immunity is well established. (17–19) However, Treg depletion is still under investigation and limited by a lack of specificity and autoimmune adverse effects. (20, 21) It is therefore necessary to identify upstream targets that mediate Treg accumulation after RT. Previous reports have detailed mechanisms by which conventional dendritic cells (cDC) can promote tolerance and expand Treg populations. (22–24) While preclinical studies have demonstrated potential for targeting cDC signaling to enhance RT, none have been clinically adopted.
Conventional dendritic cells (cDC) are canonically divided into two lineage-based populations, type I cDC (cDC1) and type II cDC (cDC2). (25–28) cDC1 have been the focus of most RT investigations due to their specialization in CD8+ T cell cross-priming. (29, 30) However, mice deficient in cDC1 still respond to RT in some tumor models, hinting towards a mixed function and that other cross-priming populations can contribute to antitumor immunity after RT. (31, 32) cDC2 are conventionally considered primary activators of CD4+ T cells, although this portrait has recently expanded to include interferon-responsive CD8+ T cell activating populations with roles in antitumor immunity. (25, 33–35) Both cDC1 and cDC2 can acquire a regulatory program and increase immune suppression markers following tumor antigen uptake that limits antitumor immunity. (28, 36, 37) These cDC are termed ‘mature dendritic cells enriched in immunoregulatory molecules’ (mregDC) due to their coexpression of both maturation and immunoregulatory markers. (36) Importantly, the role of mregDC signaling in RT-driven Treg accumulation, immune suppression and the RT response is unknown.
In this report we identify that local RT mediates an expansion of mregDC derived from cDC1 (mregDC1) that promote an immunosuppressive microenvironment. Mechanistic studies reveal that upregulation of CCL22 in mregDC after interactions with irradiated tumor cells mediates radiation-driven Treg tumor infiltration to limit the RT response. We engineered a tumor-targeted type I interferon (IFN-I, IFNα) to simultaneously block CCL22 production after RT while facilitating cDC activation and antigen cross-presentation. (1, 8, 38, 39) Importantly, the use of IFN-I in cancer treatment has declined in recent years owing to poor efficacy and dose-limiting side effects. (40–42) To overcome this we conjugated IFNα with an anti-epidermal growth factor receptor antibody (αEGFR-IFNα). As a prototype molecule, αEGFR-IFNα can target IFN-I to the microenvironment of EGFR-expressing cancers. (40, 43–48) αEGFR-IFNα reshaped tumor cDC populations, suppressed Treg infiltration and drove an effector CD8+ T cell response in combination with RT. Our findings identify an unknown role of the cDC1-CCL22-Treg axis in RT-mediated immune suppression and provide a novel therapeutic strategy for enhancing the response to RT.
MATERIALS AND METHODS
Mice, Cell Lines, and Reagents
Six- to eight-week old C57BL/6J WT mice were purchased from Envigo. Zbtb46cre (B6.Cg-Zbtb46tm3.1(cre)Mnz/J, RRID:IMSR_JAX:028538), RosaYFP (B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J, RRID:IMSR_JAX:006148), and OT-I CD8+ T cell receptor (TCR)-Tg (C57BL/6-Tg(TcraTcrb)1100Mjb/J, RRID:IMSR_JAX:003831) mice were purchased from Jackson Labs. Batf3−/− mice were graciously donated from the Kline Lab at the University of Chicago. Mice were maintained under specific pathogen-free conditions and utilized according to a protocol approved by the Institutional Animal Care and Use Committee of the University of Chicago. The MC38 (RRID:CVCL_B288) tumor cell line was provided by Dr. Yang-xin Fu. B16F10 (RRID:CVCL_0159) and B16F1 (RRID:CVCL_0158) cell lines were acquired from American Type Culture Collection (ATCC). All cell lines were authenticated and tested free of murine pathogens. B16-OVA, B16-EGFR, B16-EGFR-OVA and MC38-EGFR were previously described. (48) Cell lines were authenticated and tested for mouse pathogens by IDEXX (Westbrook, Maine). All cells were cultured in 5% CO2 and maintained in vitro in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich), 100 U/mL penicillin, 100 μg/mL streptomycin and 0.1 mmol/L Minimum Essential Medium nonessential amino acids. αCCL22 (MDC monoclonal antibody) was purchased from Invitrogen (158132) (MA5-23780, RRID:AB_2609986). αCD8 was purchased from BioXcell (BE0004-1, RRID:AB_1107671). The SiteClick Antibody Labeling Kit (R-PE, S10467) was purchased from ThermoFisher. Anti-mouse CTLA-4 (CD152) clone 9H10 was purchased from BioXcell (BE0131, RRID:AB_10950184).
Pacific blue-anti-CD45 (103126, RRID:AB_493535), pacific blue-anti-CD11b (101224, RRID:AB_755986), Pacific-blue-anti-CD80 (104724, RRID:AB_2075999), BV421-anti-I-A/I-E (107631, RRID:AB_10900075), BV510-anti-CD19 (115545, RRID:AB_2562136), BV510-anti-CD86 (105039, RRID:AB_2562370), BV570-anti-CD4 (100541, RRID:AB_10897943), BV570-anti-NK1.1 (108733, RRID:AB_10896952), BV605-anti-CCR7 (120125, RRID:AB_2715777), BV605-anti-CD3 (100351, RRID:AB_2565842), BV605-anti-CD11c (117333, RRID:AB_11204262), BV650-anti-XCR1 (148220, RRID:AB_2566410), BV711-anti-CD64 (139311, RRID:AB_2563846), BV750-anti-CD3 (100249, RRID:AB_2734148), BV785-anti-Ly6C (128041, RRID:AB_2565852), fluorescein isothiocyanate (FITC)-anti-CD45 (103108, RRID:AB_312973), FITC-anti-CD25 (101907, RRID:AB_961210), FITC-anti-Ly6C (128006, RRID:AB_1186135), Alexa Fluor 488-anti-CD8a (100723, RRID:AB_389304), AF488-anti-FceRIa (MAR-1, 134330, RRID:AB_2687239), PerCP-Cy5.5-anti-Bcl6 (358508, RRID:AB_2566191), PerCP/Cy5.5-Ly6G (127616, RRID:AB_1877271), PerCP/Cy5.5-anti-F4/80 (123128, RRID:AB_893484), PE-anti-H2Kb-SIINFEKL (141603, RRID:AB_10897938), PE-anti-CD11b (101208, RRID:AB_312791), PE-anti-XCR1 (148203, RRID:AB_2563842), PE-Dazzle594-anti-CD103 (121430, RRID:AB_2566493), PE/Cy5-anti-CD86 (105016, RRID:AB_493602), PE/Cy5-anti-CD11c (117316, RRID:AB_493566), PE/Cy7-anti-CD11c (117318, RRID:AB_493568), PE/Cy7-anti-Ly6C (128017, RRID:AB_1732093), PE/Fire810-anti-MHCII (107667, RRID:AB_2894690), APC-anti-CD11c (117310, RRID:AB_313779), APC-anti-PD-L1 (124312, RRID:AB_10612741), APC-anti-CD4 (100412, RRID:AB_312697), AF700-anti-CD206 (141734, RRID:AB_2629637), AF700-anti-CD11b (101222, RRID:AB_493705), APC/Cy7-anti-CD172a (144017, RRID:AB_2629557), BV510-anti-CD24 (101831, RRID:AB_2563894), and Zombie NIR Fixable dye (423105) were purchased from BioLegend. BUV395-anti-Ly6G (563978, RRID:AB_2716852), BUV496-anti-CD24 (612953, RRID:AB_2870229), BUV563-anti-NK1.1 (741233, RRID:AB_2870785), BUV737-anti-CD8a (564297, RRID:AB_2722580), BUV805-anti-F4/80 (749282, RRID:AB_2873657), BV421-anti-CD40 (562846, RRID:AB_2734767), BV421-anti-FcεR1α (MAR-1, 750852, RRID:AB_2874960) and BV605-anti-I-A/I-E (MHCII, 563413, RRID:AB_2738190) were purchased from BD Biosciences. PerCP-eFluor710-anti-PD-1H (46-5919-82, RRID:AB_2573811), PE-anti-FOXP3 (12-5773-82, RRID:AB_465936), PE-anti-H2Kb-SIINFEKL (12-5743-82, RRID:AB_925774) were purchased from eBioscience. AF532-anti-CD45 (58-0451-82, RRID:AB_11218871), PerCP-eFluor710-anti-B220 (46-0452-82, RRID:AB_10717389), PE/Cy7-anti-Axl (25-1084-80, RRID:AB_2734851) were purchased from Invitrogen. eBioscience™ Foxp3/Transcription Factor Staining Buffer Set was purchased from Invitrogen (00-5523-00). SIINFEKL was purchased from Invivogen (vac-sin). Mouse MDC (CCL22) ELISA kit was purchased from Abcam (ab204525). EasySep™ Mouse CD11c Positive Selection Kit II (18780), Mouse Pan-DC Enrichment Kit II (19863), Mouse CD8+ T Cell Isolation Kit (19853), and Mouse CD8a Positive Selection Kit II (18953) were purchased from StemCell Technologies. Mouse IFNγ ELISPOT Set was purchased from BD Biosciences (551083). All primers were purchased from IDT.
Production of the αEGFR-IFNα Fusion Protein
The heterodimeric Fc variant KiHss-AkKh platform was described previously. (49) The Fab fragment of Cetuximab (anti-human EGFR) was fused with the knob variant Fc region in the pEE12.4 vector (Lonza), and the murine IFNa4 was fused with the hold variant Fc region via a GGG linker in the pEE6.4 vector (Lonza). The anti-EGFR-IFNa4 heterodimer was generated by transient co-transfection of two plasmids into FreeStyle293-F cells. The supernatant containing the fusion protein was purified using CaptivA® Protein A Affinity Resin according to the manufacturer’s manual (Repligen). Heterogeneity was confirmed by SDS-PAGE and purity evaluated by SEC-HPLC.
Tumor Growth and Treatments
Tumors were established in mice by subcutaneous injection of 1 × 106 tumor cells in 0.1 mL PBS into the hind flanks. Tumors were irradiated 10–11 d after inoculation with 20 Gy while the rest of the body was shielded by lead. For tumor rechallenge, cured or naïve mice were rechallenged with 1 × 106 tumor cells. Tumor growth was monitored by measuring the length, width, and depth and volume calculated as one half the length x width x depth. For αCTLA4 treatment, 200 μg/0.1 mL PBS was given intraperitoneal (i.p.) on day 10 with RT and day 16. αCCL22 (4 μg/mLtumor) was given every other day starting day 11 for 2 weeks. αEGFR-IFNα was injected intratumoral on day 10 (10–20 μg/0.02 mL PBS) and day 14 (5–10 μg/0.02 mL PBS). αCD8 200 μg/0.1 mL PBS was administered by intraperitoneal injection on the same day as RT and again 3 days later.
RNA Sequencing and Bioinformatic Analysis
Samples for scRNA-seq of CD45+ tumor immune cells were obtained from pooled MC38 tumors in WT mice with or without treatment with 15 Gy as described previously. (16) Samples were stained using ZombieRed™ dye for 30 min and surface stained for mouse CD45 for 20 min. ZombieRed− CD45+ single cells were sorted and used for library construction. Library construction was performed using Chromium Next GEM Single Cell 3’ GEM, Library & Gel Bead Kit v3.1 (Cat: 1000128) purchased from 10X Genomics according to the manufacturer’s instructions. Target cell recovery for each library was 8.000 and sequencing was performed on an Illumina HiSeq X Ten platform.
10X Genomics Cell Ranger (v6.0.1, RRID:SCR_023221) was used to process raw scRNA-seq data into FASTQ files with “cellranger mkfastq” function, align the reads to the mouse reference genome (mm10, GENCODE vM23/Ensembl 98 released on July 7, 2020 from 10X Genomics) and count the unique molecular identifier (UMI) with “cellranger count” function. Low-quality cells were discarded if (1) the number of expressed genes was smaller than 200; (2) the proportion of mitochondrial gene expression were larger than 25%. We further identified and removed potential doublets by using DoubletFinder (v2.0.3, RRID:SCR_018771) assuming 6% doublet formation rate. The processed whole gene expression matrix was then fed to Seurat (v4.0.6, RRID:SCR_016341) for downstream analyses.
Briefly, the UMI count matrix from untreated and RT samples were integrated by Seurat::IntegrateData method after normalized with ‘SCT’ method using 3,000 highly variable feature genes. Clustering analysis was performed using the first 40 principal components for constructing the shared nearest neighbor (SNN) graph with resolution as 0.7. Later, marker genes were called by Seurat::FindAllMarkers with log2 fold change >= 1. Identified clusters were annotated by scClassify (v1.2.0) (50) based on cell types hierarchies constructed from reference datasets (E-MTAB-8832, CD45+ immune cells sorted from MC38 tumor bearing C57BL/6 mice) (51) and labeled using the selected marker gene. To validate the annotation of the DC cells, we scored each cell in our dataset using the Seurat::AddModuleScore function using expression of cDC feature genes (H2-Ab1, Flt3 and Itgax) as described in Duong, et al. (35) Dendritic cells in the two clusters were extracted and further clustered into sub clusters and annotated using the same clustering and annotating method. The dendritic cell subtype score was calculated as the fraction of RNA in a cell belonging to genes in the gene list for the mregDC, DC1 and DC2 feature gene list described in Maier, et al. (36) The scRNA-seq dataset has been deposited in the Gene Expression Omnibus (GEO) under the accession number GSE206387.
For bulk RNA-seq Live CD45+ CD11c+ cells were flow sorted from tumors and mixed with Trizol for RNA extraction. The RNA library for sequencing was constructed using SMARTer® Stranded Total RNA-Seq Kit v2 - Pico Input Mammalian (Takara Bio) and sequencing performed at the University of Chicago Genomics Facility on an Illumina NovaSEQ machine in pair-read mode with 100 bp per read. Raw sequencing and processed data is deposited within GEO under the accession number GSE262864. RNA sequencing analysis was performed using the Galaxy platform (RRID:SCR_006281). Raw data quality control was done using FastQC, and all samples were included in the analysis. In brief, trimming was performed using Trimmomatic. Sequenced reads were aligned to the mouse reference genome (mm10) using RNA STAR (RRID:SCR_004463) and quantified by featureCounts (RRID:SCR_012919). Count data was further analyzed for differential expression using DESeq2 (RRID:SCR_015687). Gene ontology enrichment analysis was performed using ShinyGo (RRID:SCR_019213) on genes with increased expression, a Log2 fold change > 2, and p value < 0.05.
Flow Cytometry
Single cell suspensions were prepared as previously described. (52) Single cells from tumors were harvested by digestion using 1 mg/mL collagenase IV (Sigma-Aldrich) and 200 μg/mL DNaseI (Sigma-Aldrich) at 37°C for 30–60 minutes. Blocking was accomplished with anti-Fc receptor (2.4G2, Leinco C381, RRID:AB_2737484) prior to cell staining. Cells were stained according to manufacturer recommendations. Surface and live/dead staining was performed in PBS for 45 minutes followed by 2 consecutive washes and fixation according to the eBioscience Foxp3/Transcription Factor Staining Buffer Set (00-5523-00). FoxP3 staining was performed in permeabilization buffer for 45 minutes at room temperature followed by 2 consecutive washes according to the FoxP3 staining protocol (ThermoFisher, 00-5523-00). CCL22-PE was prepared using the SiteClick Antibody Labeling Kit for PE (ThermoFisher) and αCCL22 according to the manufacturer instructions. CCL22-PE staining was performed using the ThermoFisher protocol for cytoplasmic proteins. Cell suspensions were analyzed on a Fortessa (BD Biosciences), Attune (Invitrogen), or Aurora (Cytek) flow cytometer. Cell sorting was performed using a AriaIII and BC FACSAria Fusion (BD Biosciences). Data was analyzed with OMIQ software from Dotmatics.
ELISPOT Assay
For CD8+ T cell functional assays, CD8+T cells were isolated from tumors using an EasySep™ Mouse CD8a Positive Selection Kit (STEMCELL Technologies) 7 d after treatment initiation. 2–4 × 105 CD8+ T cells were stimulated with 1 μg/mL SIINFEKL for 48 h. ELISPOT assays were performed to detect cytokine spots of IFNγ according to the product protocol (Millipore). In brief, a 96 well HTS-IP plate was precoated with an anti-IFNγ antibody at a 1:250 dilution and incubated overnight at 4°C. Cells were removed and biotinylated anti-IFNγ antibody (2 mg/mL) at a 1:250 fold dilution was added and incubated for 1–2 h at room temperature. Avidin-horseradish peroxidase in a 1:1000 dilution was added to the plate and incubated for 1 h at room temperature. Spots were developed according to the manufacturer’s instructions (BD) and calculated using an ELISPOT plate reader.
Ex Vivo Dendritic Cell Culture
Spleens were isolated and single cell suspensions prepared from 8–12 week old Zbtb46creRosaYFP mice as described for flow cytometry. cDC were enriched using an pan-DC enrichment kit according to the manufacturer’s instructions (STEMCELL Technologies). Enriched cell suspensions were stained for αCD24 and αCD172a and immediately sorted based on YFP and stains as shown in the representative gating strategy. B16 tumor cells were either untreated or treated with 40 Gy radiation the day prior to plating. cDC were plated at a 1:1 ratio with tumor cells in 96-well cell culture plates in 150 μL RPMI 1640 (Gibco) media supplemented with 10% FBS, penicillin and streptomycin. Cells collected by trypsinization, incubated with eBioscience Protein Transport Inhibitor Cocktail (00-4980-03) for 6 hours in supplemented RPMI at 37°C and stained for CD45 and CCL22 according to the ThermoFisher protocol for cytoplasmic protein staining. PE staining in CD45+ YFP+ cells was quantified and analyzed using OMIQ.
Statistical Analysis
For tumor growth measurements, flow cytometry, gene expression, Kaplan-Meier analysis, and protein measurements descriptive statistics were analyzed using Prism software (version 7.0, GraphPad). Kaplan-Meier survival analysis was compared using the log-rank (Mantel-Cox) test. All other data was tested using either two-way ANOVA, Student’s t-test, or one-way ANOVA as described in the corresponding figures. R was used for analysis of public datasets. The human homolog of DC_Ccl22 was generated using the Mouse Genome Informatics Database from The Jackson Laboratory. The resulting gene list was: CACNB3, CCR7, FSCN1, TBC1D4, TMEM123, SERPINB6, CCL5, CD63, SAMSN1, RAMP3, MMP25, MARCKS, FABP5, FABP5P3, CCL22, STAT4, SERPINB9, ROGDI, SOCS2, MARCKSL1, CD200, CALM1, ISCU, RELB, BCL2A1, BASP1, TRAF1, MAP4K4, IL4I1, GYG1, PCGF5, PDLIM4, GADD45B, ANXA3, BIRC2, ARL5C, KTN1, RPS27L, GLIPR2, NET1, UAP1, ID2, ZMYND15, TXNDC17, GNB4, EPSTI1. Data from the Farren, et al. dataset was used from the Gene Expression Omnibus dataset under accession GSE129492. (53) TCGA data was acquired and analyzed in part using the Xena Platform. (54) For analysis of gene signatures in survival and correlation each cohort gene expression was divided into tertiles. Samples in the lowest and highest tertiles received a score of −1 and 1, respectively. Scores were added for each sample and the lowest quartile and highest tertile isolated at ‘Low’ and ‘High’ gene expression signature, respectively. Survival data from these two groups were then compared. Data analysis of TCGA and public datasets was performed using R. Sample sizes were determined based on previous reports and all animals were randomly divided into treatment groups based on tumor volume. The number of biological replicates for all bar graphs are represented by individual data points. All tumor growth/survival and flow cytometry experiments are representative of at least 2 independent experiments. For all tumor growth and survival studies a minimum of 5 mice per group were used. All experiments were unblind. All data presented as mean ± standard error unless otherwise noted. n.s. (not significant), *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
Study Approval
Mice were maintained under specific pathogen-free conditions and utilized according to protocol approved by the Institutional Animal Care and Use Committee of the University of Chicago.
Data and materials availability:
The scRNA-seq dataset has been deposited in the Gene Expression Omnibus (GEO) under the accession number GSE206387. Raw bulk RNA sequencing and processed data is deposited within GEO under the accession number GSE262864. All other data is available at request from R.R.W.
RESULTS
Local irradiation expands tumor mregDC
We previously performed single-cell RNA sequencing (scRNA-seq) on CD45+ cells isolated from murine MC38 colorectal adenocarcinoma subcutaneous tumors 4 days after treatment with 15 Gy to explore changes in suppressive myeloid populations (Supplementary Figure S1A). (16) Doses of 15–20 Gy were utilized since they have demonstrated CD8+ T cell-dependent antitumor immunity and are included in various stereotactic ablative regimens from clinical studies. (55–58) A timepoint of 4 days was chosen as we observe significant tumor myeloid cell infiltration and cDC activation 3–5 days after ablative regimens. (8, 15, 16, 59, 60) Twenty-four populations were identified using unbiased clustering of all CD45+ tumor infiltrates based on canonical immune signatures. Both unbiased assignment and mapping a cDC signature score based on expression of H2-Ab1, Flt3 and Itgax (35) revealed 2 cDC clusters annotated by signature gene expression (DC_Ccl22 and DC_Atox1, Supplementary Figure S1A–B). DC_Atox1 exhibited characteristic expression of both cDC1 and cDC2 markers. Instead, DC_Ccl22 exhibited an upregulation of markers of maturation, migration and regulation, suggesting an activated mregDC phenotype. Scoring for cDC1, cDC2 and mregDC as described in Maier, et al. revealed an enriched mregDC score in the DC_Ccl22 cluster (Supplementary Figure S1C). To examine changes in cDC lineage-based populations, we isolated cDC and identified 3 canonical populations derived from DC_Atox1: DC_Rab7b (cDC1), DC_Ly6a (cDC2), and DC_Cd7 (plasmacytoid dendritic cells, pDC), as well as a distinct separate population of mregDC (DC_Ccl22) (Figure 1A–C).
Figure 1. Local irradiation expands a population of mature cDC enriched in regulatory markers within the tumor.

A, Heatmap of top 20 differentially expressed genes for each dendritic cell cluster identified. MC38 tumors were treated with 15 Gy ionizing radiation and live CD45+ cells sorted 4 days after treatment for scRNA-seq. B, Dendritic cell clusters identified from DC_Ccl22 and DC_Atox1 revealed 4 distinct dendritic cell populations (most closely resembling): DC_Ccl22 (mregDC), DC_Ly6a (cDC2), DC_Cd7 (pDC), and DC_Rab7b (cDC1). C, Stratification of cDC clusters using mregDC signature scores generated from gene lists provided in Maier, et al. D, Changes in cDC cell clusters after 15 Gy irradiation as a percentage of total cDC.
The DC_Ccl22 cluster shared a high similarity with mregDC reported in Maier, et al. (36), including an upregulation of genes involved in immune regulation (Cd274, Socs2, Aldh1a2, Cd200, and Ccl22), a type II helper T cell response (Il4i1, Ccl22), cDC maturation (Relb, Cd80, and Cd86), and cell migration (Ccr7, Myo1G, Fscn1, and Marcks) (Figure 1A, C and Supplementary Figure S1D). Among cDC clusters, DC_Ccl22 exhibited the greatest proportionate increase after RT and upregulation of immune suppression markers (Cd274, Cd200 and Socs2), suggesting that they may contribute to RT-driven immune suppression (Figure 1D and Supplementary Figure S1D). Moreover, among the top genes differentially expressed in DC_Ccl22, the known Treg-recruiting chemokine CCL22 was exclusively restricted to the DC_Ccl22 cluster (Supplementary Figure S1E). We developed a 46-gene human homolog signature using the top differentially expressed genes in DC_Ccl22 to examine an association with cancer patient outcomes. A higher Human Ccl22 Signature Score was associated with worse survival in The Cancer Genome Atlas (TCGA) Pan-Cancer Project (PANCAN, Supplementary Figure S1F).
Single cell RNA sequencing alone was unable to distinguish mregDC derived from each cDC lineage (mregDC1 and mregDC2), similar to previous studies which utilized surface marker labeling to distinguish these populations. (36) We performed spectral flow cytometry of radiosensitive MC38 and radioresistant B16 murine melanoma expressing the model tumor antigen ovalbumin (B16-OVA) to characterize changes in tumor cDC populations after treatment with 20 Gy (Figure 2 and Supplementary Figure S2–3). B16-OVA was also utilized to identify MHCI-SIINFEKL+ cDC populations (processed model OVA tumor antigen presented on MHCI). cDC cells were gated as Live CD45+ F4/80− MHCIIHI CD11cHI and either CD103+ (cDC1) or CD11b+ (cDC2) (Supplementary Figure S2A). cDC were plotted by UMAP and unsupervised clustering performed with FlowSOM. (61) In both tumor models unsupervised clustering revealed 4 tumor cDC populations (Figure 2A–B). Clusters were distinguished by expression of canonical lineage markers for cDC1 (CD24, XCR1 and CD103) and cDC2 (CD11b, CD172a), as well as maturation markers for mregDC1 and mregDC2 (lineage markers plus CCR7 and CD40) (Figure 2A, C and Supplementary Figure S2B–E). CCR7 and CD40 were characteristically upregulated in the DC_Ccl22 mregDC cluster and have been previously reported as mregDC markers. (36) Similarly, both mregDC1 and mregDC2 exhibited greater staining for MHCI-SIINFEKL, confirming mregDC as the major cDC antigen presenting population after RT (Figure 2C). Both mregDC1 and mregDC2 also had greater staining for the cell surface coinhibitory molecules PD-L1 and VISTA, a potent suppressor of the early stages of T cell activation, compared to their resting state cDC1 and cDC2 counterparts, respectively (Supplementary Figure S2F). (62–65) Herein, lineage-based resting populations are described as cDC1 and cDC2, whereas mregDC1 and mregDC2 contain both lineage markers and expression of mregDC markers.
Figure 2. Radiation increases tumor mregDC1.

A, UMAP of cDC1 and cDC2 from MC38 tumors (pooled from No Treatment, 3, 5 and 7 days after 20 Gy) using spectral flow cytometry. Unsupervised clustering with FlowSOM reveals 4 subpopulations of tumor cDC, including both resting cDC lineages (cDC1 and cDC2) and mregDC (mregDC1 and mregDC2). Color scale corresponds to either high (yellow) or low (dark blue) marker expression as measured by normalized MFI (scaled mean). B, UMAP of cDC from B16-OVA tumors (pooled from No Treatment and 5 days after 20 Gy) using spectral flow cytometry. Unsupervised clustering (FlowSOM) revealed 4 subpopulations of tumor cDC with similar marker expression patterns as observed in panel A. C, mregDC1 and mregDC2 clusters exhibit increased surface staining for CCR7 and H2Kb-SIINFEKL. D, Changes in cDC populations identified in panel b from B16-OVA tumors 5 days after treatment with 20 Gy. Data presented as mean ± SEM. Panel C: Mixed-effects analysis with Tukey’s multiple comparisons. Panel D: Unpaired t-test. ns: not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
In both MC38 and B16-OVA tumors, cDC1 and mregDC1 were significantly increased 5 days after 20 Gy (Supplementary Figure S3A–B and Figure 2D, respectively). In contrast, while a trend towards an increase in cDC2 and mregDC2 was observed in B16-OVA tumors, this was not the case in MC38. These observations were consistent with conventional gating approaches as both CCR7+ cDC1 and H2Kb-SIINFEKL+ cDC1 were significantly elevated 5 days after 20 Gy in B16-OVA tumors (Supplementary Figure S3C–D). Taken together with scRNA-seq findings, these observations show that RT increases a population of mregDC1 with increased regulatory markers and immune suppressive potential.
mregDC production of CCL22 drives Treg immune suppression after RT
The expansion of mregDC1 and their enrichment in regulatory markers suggested that they contribute to immune suppression after RT. The Treg-recruiting chemokine CCL22 was among the top genes enriched in mregDC (Log2FC 3.7, Padjusted = 9 × 10−74; Figure 1A). Moreover, expression of Ccl22 among all CD45+ infiltrating tumor cells was completely restricted to the DC_Ccl22 cluster (Supplementary Figure S1E). Treg tumor infiltration and their role suppression of RT efficacy is well established. (12–14, 17) Consistent with this, expression of the Treg marker FOXP3 among pancreatic cancer patient tumor samples after RT was significantly increased in the dataset reported in Farren, et al (Supplementary Figure S4A). (53) We also observed a positive correlation between the Human DC_Ccl22 Homolog Score and both FOXP3 and CCL22 expression across the majority of TCGA solid tumor cohorts (Supplementary Figure S4B and Supplementary Figure S5, respectively).
Treg increased in both B16 and MC38 tumors 5 days after 20 Gy, with B16 exhibiting a greater overall increase (Figure 3A and Supplementary Figure S6A–B). Administration of a CCL22 neutralizing antibody (αCCL22) significantly decreased tumor Treg 5 d after 20 Gy in both models (Figure 3B and Supplementary Figure S6B). For further mechanistic studies we focused on B16 tumors given their more radioresistant phenotype and larger magnitude of Treg accumulation after RT. (66) Tumor levels of CCL22 protein were significantly elevated by day 3 after RT (Figure 3C). Previous studies suggest that mregDC1 exhibit increased Ccl22 expression in murine lung tumors compared with mregDC2. (36) To determine the contribution of cDC lineages in CCL22 production after RT we isolated splenic cDC from Zbtb46cre RosaYFP mice in which cre recombinase under control of the Zbtb46 promotor expressed in cDC cleaves a loxP-flanked STOP sequence allowing expression of the fluorescent protein YFP in cDC. (67) Splenic cDC1 (YFP+ CD24+) and cDC2 (YFP+ CD172a+) were sorted, cocultured with either untreated or irradiated B16 tumor cells for 18 h, and then analyzed by flow cytometry for CCL22 expression (Figure 3D and Supplementary Figure S6C). Although cDC1 exhibited a higher baseline CCL22 staining compared with cDC2, coculture with irradiated tumor cells only induced CCL22 in cDC1.
Figure 3. Regulatory T cell-mediated immune suppression after radiotherapy is driven by mregDC1 production of CCL22.
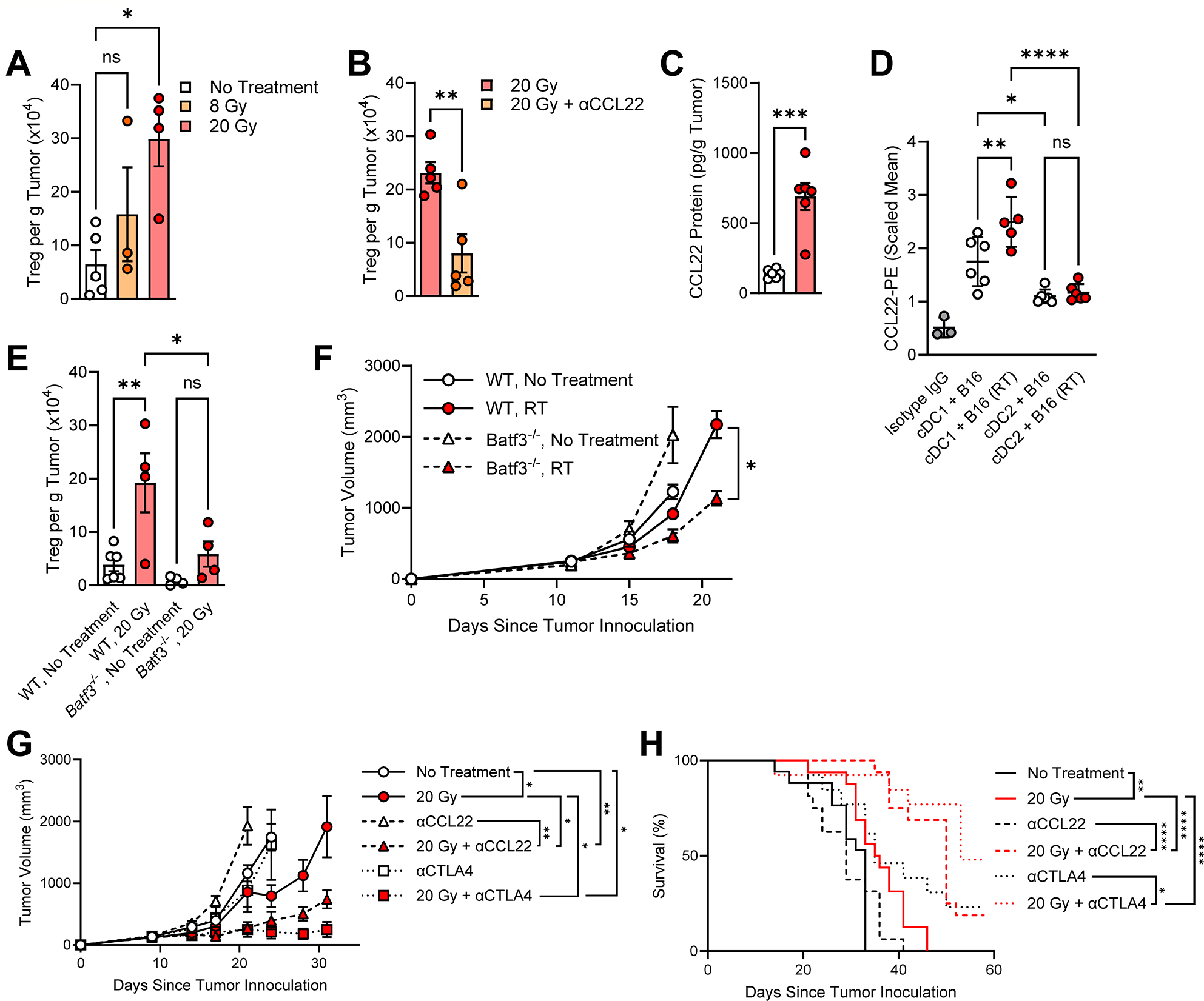
A, Tumor Treg numbers in B16 tumors 5 days after treatment with either 8 Gy or 20 Gy as determined by flow cytometry. B, Treg in B16 tumors 5 days after treatment with 20 Gy in combination with αCCL22 (4 μg/mLtumor was administered by intratumoral injection 1 day and 4 days after a single dose of 20 Gy). C, CCL22 protein in B16 tumors 3 days after treatment with 20 Gy. D, Quantification of intracellular CCL22 staining in splenic YFP+ CD24+ cDC1 or YFP+ CD172a+ cDC2 isolated from Zbtb46cre RosaYFP mice cocultured in the presence of either non-irradiated or irradiated B16 tumor cells for 18 h. E, Treg in B16 tumors grown in either WT or Batf3−/− mice isolated 5 days after treatment with 20 Gy analyzed by flow cytometry. F, Tumor growth of B16F1 in either WT or Batf3−/− mice. 20 Gy was given on day 11. G-H, Tumor growth and survival of mice bearing B16 tumors treated with 20 Gy in combination with either αCCL22 or αCTLA4 clone 9H10. αCCL22 was administered intratumoral at a concentration of 4 μg/mLtumor as determined by caliper measurement 3 days per week for 2 weeks beginning 1 day after RT (day 10). αCTLA4 9H10 was administered intraperitoneal (IP) as 200 μg on day 10 with RT and day 16. For image clarity, significant survival comparisons between No Treatment vs. 20 Gy + αCCL22 (P < 0.01) and No Treatment vs. 20 Gy + αCTLA4 (P < 0.01) were not shown on the figure. Data presented as mean ± SEM. Panel C: Mixed-effects analysis with Tukey’s multiple comparisons. Panels A, D, E: One-way ANOVA with Tukey’s multiple comparisons. Panels B, C, F (day 21): Unpaired t-test. Panel G (day 24, all groups): Mixed-effects model with Geisser-Greenhouse correction and Tukey’s multiple comparisons at day. Panel G (day 31, remaining groups): One-way ANOVA with Dunnett’s multiple comparisons. Panel H: Log-rank test. ns: not significant, *P < 0.05, **P < 0.01, ****P < 0.0001.
To examine the role of cDC1 on Treg accumulation after RT we used flow cytometry to analyze Treg populations in Batf3−/− mice, as they are deficient in cDC1 and continue to be absent from tumors after irradiation (Supplementary Figure S7A). B16 tumors in Batf3−/− mice had significantly less Treg accumulation 5 d after 20 Gy compared to tumors in wild type (WT) mice (Figure 3E). To determine if cDC1 contribute to radioresistance we examined the tumor growth of B16F1 melanoma, a highly aggressive radioresistant cell line. (66) Despite a faster baseline tumor growth in Batf3−/− mice, irradiated tumors responded significantly better to 20 Gy in Batf3−/− mice compared to those in WT (Figure 3F). It is important to note that Batf3−/− mice have additional alterations in immune cells that limit the use of this model. However, it has been previously demonstrated that Batf3−/− mice exhibit favorable CD4+ T cell differentiation into Treg both in vitro and in the murine gut. (68) Taken together, these findings suggest that cDC1 is a major contributor to Treg accumulation and radioresistance through production of CCL22.
We hypothesized that perturbing the cDC1-CCL22-Treg axis may hold therapeutic potential to enhance RT efficacy. Mice bearing either B16 or MC38 tumors responded significantly better to RT in the presence of αCCL22 by both tumor growth and survival (Figure 3G–H and Supplementary Figure S7C–D, respectively). To determine if radiation-recruited Treg suppress the RT response and represent a potential therapeutic target, we treated tumor bearing mice with anti-CTLA4 clone 9H10, as this clone has been shown to deplete Treg in mice. (69–71) Concurrent treatment with αCTLA4 clone 9H10 significantly enhanced the antitumor activity of local RT and prolonged survival in mice bearing B16 and MC38 tumors (Figure 3G–H and Supplementary Figure S7C–D, respectively). These results highlight the therapeutic potential of targeting the cDC1-CCL22-Treg axis to overcome RT-driven immune suppression and enhance RT efficacy, as both CCL22 blockade and Treg depletion were independently able to augment the RT response.
Tumor targeted type I interferon reshapes tumor cDC populations
Though several clinical trials are ongoing, Treg depletion is not yet standard of care in humans due to a lack of selective targets and autoimmune adverse effects. (20, 21) IFN-I, a potent immune stimulator, has been shown to both suppress cDC CCL22 expression (72, 73) and drive cDC-mediated antitumor immunity after RT via enhanced T cell priming. (8) Indeed, the IFN-I IFNβ abrogated cDC1 CCL22 induction upon coculture with irradiated tumor cells (Figure 4A). Therefore, we hypothesized that IFN-I may relieve RT-driven cDC1-CCL22-Treg immune suppression and simultaneously enhance the priming of an antitumor CD8+ T cell response after RT.
Figure 4. Tumor-targeted type I interferon reprograms tumor myeloid populations after radiotherapy away from immune suppression.

A, Quantification of intracellular CCL22 staining in splenic cDC1 isolated from Zbtb46cre RosaYFP mice and cocultured with irradiated B16 cells in the presence of IFNβ 10 μg/mL for 18 h. B, Structure of the αEGFR-IFNα fusion protein. C, Biological pathways enriched in CD11c+ cells isolated from tumors after treatment with the combination of 20 Gy and αEGFR-IFNα compared with RT alone. Mice were given 20 μg αEGFR-IFNα intratumoral on same day as RT and 10 μg 4 days later. Tumors were collected 7 days after RT and cells were sorted as Live CD45+ CD11c+ for RNA-sequencing. Chart produced using ShinyGO 0.76.1. D, Normalized gene expression in CD11c+ cells of the DC_Ccl22 cluster signature genes identified in Figure 1A after concurrent treatment with αEGFR-IFNα and RT. E, Relative expression of Ccl22 in Live CD45+ CD11c+ Ly6C− cells isolated from B16-EGFR tumors 3 d after local RT and 20 μg intratumoral αEGFR-IFNα. Data presented as mean ± SEM. Panels A and E: One-way ANOVA with Tukey’s multiple comparisons. ns: not significant, *P < 0.05, **P < 0.01.
Though long identified for its role in antitumor immunity, exogenous IFN-I use in cancer treatment has declined due to limited efficacy, poor pharmacokinetic properties, and dose limiting immune-mediated systemic side effects. (40–42) We previously developed an antibody-based tumor targeted IFN-I that is preferentially retained in the tumor microenvironment, improving efficacy and avoiding off-target systemic effects. (48) Based on this, we constructed a fusion protein consisting of the IFN-I IFNα4 linked to an antibody against human epidermal growth factor receptor (αEGFR), resulting in a heterodimeric αEGFR-IFNα fusion protein (Figure 4B). EGFR is a commonly overexpressed oncogenic membrane protein in various human cancers. (47) We hypothesized that αEGFR-IFNα can target IFN-I to the tumor microenvironment to increase potency, simultaneously inhibiting CCL22 production and driving an antitumor T cell response.
We performed bulk RNA-sequencing of live CD45+ CD11c+ cells isolated from B16 tumors expressing human EGFR (B16-EGFR) to investigate the effect of αEGFR-IFNα on antigen presenting myeloid cell populations after RT (Figure 4C–D and Supplementary Figure S8A–D). Mice received an intratumoral 20 μg of αEGFR-IFNα on the same day as RT and 10 μg 4 days later. Tumors were collected 3 days after the second dose (day 7 after RT). Compared to 20 Gy alone, cells isolated from tumors treated with 20 Gy and αEGFR-IFNα exhibited an upregulation in transcriptional programs associated with a response to IFN-I and positive regulation of innate immune activation (Figure 4C). In contrast, biological processes related to cell substrate adhesion, angiogenesis, cell migration and motility were significantly downregulated following the combination treatment (Supplementary Figure S8A). While the DC_Ccl22 cluster signature was enriched in tumor CD11c+ cells after 20 Gy alone, many of these transcripts were down regulated after treatment with both 20 Gy and αEGFR-IFNα (Figure 4D). Although tumor CD11c+ cells contain myeloid populations other than cDC, including monocytes and macrophages subsets, we detected a trend towards an overall decrease in Ccl22 as well as other Treg-recruiting chemokines (Ccl1, Ccl17, Ccl20 and Ccl28) in tumors treated with αEGFR-IFNα and RT (Supplementary Figure S8B–D). We measured Ccl22 mRNA by qPCR in live CD45+ CD11c+ Ly6C− cells isolated from B16-EGFR tumors 3 d after 20 Gy. Expression of Ccl22 was 14-fold lower in the combination treatment group compared to irradiated tumors (Figure 4E). Similarly, CCL22+ cDC1 were decreased to near-undetectable levels in B16 tumors 3 days after treatment with RT and αEGFR-IFNα as measured by flow cytometry (Supplementary Figure S8E).
We examined changes in tumor cDC populations in the presence of αEGFR-IFNα after treatment with 20 Gy by flow cytometry (Figure 5). All cDC populations were significantly decreased in B16-EGFR tumors 5 d after irradiation in mice also receiving αEGFR-IFNα: cDC1, CCR7+ cDC1 (mregDC1), cDC2 and CCR7+ cDC2 (mregDC2) (Figure 5A). Although unsupervised clustering revealed 2 populations of cDC2, due to the low abundance of cDC1 and mregDC1 following αEGFR-IFNα treatment, clustering of pooled samples failed to distinguish multiple populations of cDC1 (Figure 5B–C). However, unsupervised FlowSOM clustering of all cDC revealed the expansion of a new population of cDC2 particularly enriched in tumors treated with αEGFR-IFNα (Figure 5B–D). Recent reports have detailed distinct cDC2 populations that become efficient CD8+ T cell activators in the presence of IFN-I. (34, 35) The new population of cDC2 that emerged after treatment with αEGFR-IFNα and radiation exhibited characteristic upregulation of markers for interferon-stimulated genes cDC2 (ISG+ cDC2) reported in Duong, et al., including CD80, CD86, AXL, MAR-1 and CD64 (Figure 5B). (35) Moreover, the proportion of ISG+ cDC2 significantly increased with the combination of 20 Gy and αEGFR-IFNα compared with irradiation alone (Figure 5D). We also observed an increase in the gene signature for interferon-stimulated cDC2 reported in Duong, et al. (35) and Bosteels, et al. (34) among bulk RNA-seq of tumor CD11c+ cells (Supplementary Figure S9A–B, respectively). Together, these findings demonstrate that αEGFR-IFNα reshapes tumor cDC populations after RT, decreasing CCL22+ cDC1 and inducing ISG+ cDC2.
Figure 5. Tumor-targeted type I interferon remodels the tumor cDC population and induces a population of ISG+ cDC2.

A, Changes in cDC populations by flow cytometry isolated from B16-EGFR tumors 5 days after 20 Gy as determined by conventional gating strategies. αEGFR-IFNα was administered intratumoral as 20 μg on the same day as RT. B-C, Identification of cDC cell clusters by flow cytometry 5 days after 20 Gy. αEGFR-IFNα was administered intratumoral as 20 μg on the same day as RT. Clusters were identified by unsupervised clustering with FlowSOM. Surface marker expression (B) and dimensionality reduction (C) were used to determine cDC populations and was consistent with induction of a recently described population of interferon-stimulated cDC2 (ISG+ cDC2). D, Changes in the proportions of cDC clusters identified in panel B and C across treatment groups. Data presented as mean ± SEM. Panels A and D: One-way ANOVA with Tukey’s multiple comparisons. Panel B: Two-way ANOVA with Tukey’s multiple comparisons. ns: not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
αEGFR-IFNα overcomes RT-driven Treg immunosuppression and augments an effector T cell response to improve RT efficacy
Given the decrease in myeloid Ccl22 expression, increase in ISG+ cDC2, and known effects of IFN-I on cDC activation, we hypothesized that αEGFR-IFNα could simultaneously decrease CCL22 and Treg tumor accumulation, and augment an effector T cell response after RT. Indeed, both CCL22 and Treg were significantly decreased in B16-EGFR tumors 5 d after treatment with 20 Gy and αEGFR-IFNα compared to 20 Gy alone (Figure 6A–B, respectively). Moreover, the combination of αEGFR-IFNα and 20 Gy significantly increased tumor CD8+ T cell infiltration (Figure 6C). To determine the effect of αEGFR-IFNα on the tumor antigen-specific effector CD8+ T cell response we isolated CD8+ T cells from B16-EGFR-OVA tumors 7 d after irradiation and pulsed them with the OVA peptide ex vivo. The combination treatment significantly enhanced the effector CD8+ T cell response compared with any other treatment as measured by IFNγ ELISPOT assay (Figure 6D). These observations demonstrate that αEGFR-IFNα can both simultaneously alleviate RT-driven Treg immunosuppression and augment an antitumor effector T cell response.
Figure 6. Tumor-targeted type I interferon alleviates CCL22-driven Treg immune suppression after radiotherapy to augment radiation efficacy.

A, CCL22 protein measurements in B16-EGFR tumors 3 days after treatment with 20 Gy with or without concurrent αEGFR-IFNα. Intratumoral αEGFR-IFNα was given as 20 μg on the day of RT. P = 0.06 between No Treatment and 20 Gy + αEGFR-IFNα. B, Treg as measured by flow cytometry in B16-EGFR tumors isolated 5 days after irradiation with 20 Gy. Intratumoral αEGFR-IFNα was administered as 20 μg on the day of irradiation and 10 μg four days later. C, CD8+ T cells as measured by flow cytometry in B16-EGFR tumors 7 days after 20 Gy with or without αEGFR-IFNα administered as described in panel B. D, Ex vivo IFNγ ELISPOT assay of CD8+ T cells isolated from OVA-expressing B16-EGFR tumors 7 days after local RT with or without αEGFR-IFNα and pulsed with the OVA peptide. αEGFR-IFNα administered as described in panel B. E-F, Tumor growth and survival of mice bearing B16-EGFR tumors treated with 20 Gy with or without intratumoral αEGFR-IFNα. αEGFR-IFNα was given as described in panel B. G-H, Tumor growth and survival of mice bearing B16-EGFR tumors treated with 20 Gy with or without intravenous (IV) αEGFR-IFNα. αEGFR-IFNα was given using the same schedule as in panel B. IFNα4 was given IV at a molar dose equivalent to αEGFR-IFNα using the same dosing schedule as panel B. Data presented as mean ± SEM. Panels A-D: One-way ANOVA with Tukey’s multiple comparisons. Panels E and G at day 21: Two-way ANOVA with Tukey’s multiple comparisons. Panels F and H: Log-rank test. ns: not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We treated mice bearing either B16-EGFR or MC38-EGFR tumors with intratumoral αEGFR-IFNα in combination with 20 Gy radiation (Figure 6E–F and Supplementary Figure S10A–B, respectively). In both tumor models, the combination treatment of RT and αEGFR-IFNα significantly inhibited tumor growth and prolonged survival compared to any other treatment moiety alone. Moreover, 44% of mice bearing B16-EGFR and 100% of mice bearing MC38-EGFR exhibited complete tumor regression. These mice were rechallenged by a subcutaneous reinjection of tumor cells and all mice failed to reestablish tumors (Supplementary Figure S10C–D, respectively). To validate our prototype design for systemic administration of IFN-I we compared its effectiveness after IV administration with a molar equivalent dose of unmodified IFNα4 (free IFNα4, Figure 6G–H). Mice bearing B16-EGFR tumors responded significantly better to αEGFR-IFNα compared with either RT alone or in combination with free IFNα4. Among mice treated with RT and αEGFR-IFNα IV 29% exhibited complete tumor regression at 60 days (50 days after RT).
We examined expression of immunoregulatory markers and effector T cell-recruiting chemokines among CD11c+ RNA-sequencing data (Supplementary Figure S10E). The addition of αEGFR-IFNα tended to decrease expression of multiple immunoregulatory markers, including Ptgs2, Nos2, Ido1 and Arg1, although did increase Cd274. RT and αEGFR-IFNα also increased expression of the T cell-recruiting chemokines Cxcl10 and Cxcl11. Given this and the profound increase in CD8+ T cells after the combination treatment we depleted CD8+ T cells using αCD8 and examined the effect on the RT and αEGFR-IFNα response. CD8+ T cell depletion ablated RT + αEGFR-IFNα efficacy as measured both by tumor growth and survival (Supplementary Figure S10F–G). Taken together, these findings demonstrate that αEGFR-IFNα can block Treg infiltration after RT and simultaneously mediate an effector CD8+ T cell response to generate robust antitumor responses.
DISCUSSION
There is a growing interest in expanding RT as both a local and systemic cancer treatment. One approach has been to combine RT with immunotherapy. However, responses to the combination are not always consistent and there has been limited progress in the metastatic setting. (9–11, 74–76) A growing body of evidence suggest that overcoming immune suppression due to radiation, particularly at ablative doses, is necessary to unleash antitumor immunity and poise the tumor for the radioimmunotherapy combination. (1, 15, 16, 77) Our strategy was to simultaneously alleviate radiation-driven Treg immune suppression and enhance cDC antitumor function.
We identified a targetable immunoregulatory program enriched in cDC characterized by high expression of the Treg-recruiting chemokine CCL22 after RT. These cells were analogous to mregDC tumor populations recently described. (28, 36, 37) Since we sequenced all CD45+ tumor cells, our study was not designed to detect small differences within the mregDC transcriptomic program unique to the acute inflammatory environment after radiation. Future studies aimed at elucidating steady tumor state vs. radiation-driven acute inflammation mregDC populations may help identify therapeutic strategies specific to the radiation setting. Moreover, dissecting heterogeneity within the mregDC population after RT may provide further insights into triggers underlying the development cDC-mediated immune suppression after radiation. Based on the mregDC signature, we identified for the first time CCL22 as a key targetable mediator of RT-induced Treg accumulation and immunosuppression.
Multiple therapeutic approaches aimed at blocking the cDC1-CCL22-Treg axis particularly enhanced RT efficacy. CCL22 blockade failed to delay tumor growth in non-irradiated tumors, but significantly improved RT response in both B16 and MC38 tumors. While Treg depletion with the αCTLA4 9H10 clone alone did exhibit some efficacy in MC38 tumors, this effect was not observed in B16 tumors. Notably, Treg depletion improved the RT response in both models. αCTLA4 does not deplete Treg in humans, however anti-CCR4 (CCL22 cognate receptor) antibodies can decrease Treg in humans and promote tumor immunity, suggesting a potential application in combination with RT. (70, 78) Ex vivo models demonstrated that cDC1 were the main producers of CCL22. Interestingly, B16 tumors exhibited greater overall Treg infiltration after RT and cDC1-deficient mice bearing radioresistant B16F1 tumors had an improved RT response despite faster baseline tumor growth. While our work does not exclude the importance of cDC1 in RT-induced antitumor immunity, it does highlight a likely critical role of alternative cross-presenting cells in RT. (79) Together, this evidence also suggests a tumor cell-specific mechanism for Treg-mediated radioresistance that is dependent on cDC1, and future insights may provide a strategy to better tailor cancer treatment plans.
We focused our studies on high doses (15–20 Gy) given their inclusion in stereotactic ablative regimens from clinical studies and dependence on CD8+ T cell-dependent antitumor immunity. (55–58) However, the role of dose in the cDC1-CCL22-Treg axis remains an area for future study. In our B16 model we did not observe a significant increase in Treg tumor accumulation at a moderate dose of 8 Gy. One potential explanation for this observation is that higher doses of RT (>12 Gy) induce expression of the STING activation antagonist DNA exonuclease Trex1 which impairs tumor cell IFN-I production. (80) In contrast, lower doses of RT do not result in Trex1 induction, leading to increased IFN-I production and potential suppression of cDC1 CCL22.
αEGFR-IFNα completely ablated radiation induction of CCL22 and Treg accumulation to levels below even non-treated tumors. At the same time αEGFR-IFNα significantly decreased all tumor resident cDC populations. Although it is likely that cDC activated in the presence of αEGFR-IFNα exhibited enhanced migration to the tumor draining lymph nodes, we were not able to distinguish tumor mregDC populations in the lymph due to changes in mregDC marker expression upon lymph node entry. (48) Thus, the relative contribution of direct αEGFR-IFNα Ccl22 inhibition vs. an effect from overall tumor mregDC1 decrease on Treg tumor infiltration after RT is unclear.
Although αEGFR-IFNα decreased tumor cDC populations, it strongly induced a subset of ISG+ cDC2. Untreated tumors were nearly completely devoid of ISG+ cDC2. ISG+ cDC2 were a minor cDC population after RT, whereas they made up more than half of all cDC in tumors receiving the combined treatment of RT and αEGFR-IFNα. Though many immunoregulatory markers were decreased in tumor CD11c+ cells after treatment with RT and αEGFR-IFNα, a notable exception was an upregulation in Cd274 (PD-L1). ISG+ cDC2 also exhibited significant upregulation of surface PD-L1. Dendritic cell expression of PD-L1 attenuates T-cell immunity and is believed to contribute to the response to anti-PD-L1 checkpoint blockade. (81, 82) Thus, future regimens combining RT, tumor-targeted IFN-I and αPD-L1 may have therapeutic potential.
Interestingly, interferon-stimulated cDC2 have been shown to cross-present antigen via a variety of different pathways. (34, 35) In tumors, ISG+ cDC2 can directly acquire tumor antigen-MHCI complexes from tumor cells in a process known as cross-dressing. (35) Given their induction here, as well as the dispensability of cDC1 in our radioresistant B16F1 model, it would be insightful to investigate the contribution of cross-dressing to the radiation and αEGFR-IFNα response.
Here we profiled the underlying cDC signaling after RT to identify key targetable vulnerabilities. Blocking Treg infiltration after RT either via αCCL22 or Treg depletion significantly enhanced the RT response and provided one potential therapeutic strategy. Simultaneously inhibiting CCL22 expression and inducing ISG+ cDC2 with a tumor targeted IFN-I generated the most robust antitumor immune response after RT. Our findings not only provide a novel strategy for enhancing RT response, but also highlight the potential for bifunctionally targeting the tumor cDC population in the treatment of cancer.
Supplementary Material
Translational Relevance:
Radiotherapy is used in many cancer treatment regimens and works in part by activating immunogenic tumor immunity. Combining checkpoint blockade with radiotherapy garnered excitement in preclinical studies. However, clinical results have been mostly disappointing. Radiation paradoxically induces negative regulators of tumor immunity that cause radioresistance, including regulatory T cells (Treg). Treg depletion is still under investigation and limited by a lack of specificity and autoimmune side effects. Blocking the Treg-recruiting chemokine CCL22 produced by dendritic cells (cDC) enhanced radiotherapy efficacy in mice. Although type 1 interferon inhibits cDC CCL22 production and enhances cDC T cell priming, its clinical use has gone out of favor due to limited potency. Our prototype tumor-targeted interferon simultaneously blocked cDC CCL22 production and induced a T cell response with robust antitumor activity in mice. Combined with RT, systemic administration outperformed unmodified interferon. We identify novel targets and provide a therapeutic strategy for overcoming radioresistance.
Acknowledgments:
We wish to thank R. Torres for assistance in animal studies. We thank The University of Chicago CAT Facility (RRID: SCR_017760) for their assistance with Flow Cytometry. R.R. Weichselbaum received funding from Mr. and Mrs. Vincent Foglia, The Chicago Tumor Institute, The Ludwig Cancer Research Foundation, and NIH R21CA226582, R01CA262508 and U54 CA 274291. J. Bugno received support in the form of a Clinical Therapeutics Training Grant (T32GM007019) and from the National Cancer Institute (K12CA139160). A. Piffko received support from Deutsche Forschungesgemeinschaft (DFG, German Research Foundation) – project nr. 455353745.
Footnotes
Competing interests: RRW has stock and other ownership interests with Boost Therapeutics, Immvira LLC, Reflexion Pharmaceuticals, Coordination Pharmaceuticals Inc., Magi Therapeutics, Oncosenescence, Aqualung Therapeutics Corporation, Cyntegron and PersonaDX. He has served in a consulting or advisory role for Aettis Inc., AstraZeneca, Coordination Pharmaceuticals, Genus, Merck Serono S.A., Nano Proteagen, NKGen Biotech, Shuttle Pharmaceuticals, Highlight Therapeutics, S.L., Aqualung Therapeutics Corporation. He has a patent pending entitled ‘Methods and Kits for Diagnosis and Triage of Patients with Colorectal Liver Metastases’ (PCT/US2019/028071). He has received research grant funding from Varian and Regeneron. He has received compensation including travel, accommodations or expense reimbursement from Astrazeneca, Boehringer Ingelheim and Merck Serono. C.H. has stock and other ownership interests with Accent Therapeutics, Inc. and Aferna Green, Inc. C.H. is scientific advisory board member of Aferna Green and Rona Therapeutics. The remaining authors declare no conflicts of interest.
References
- 1.Spiotto M, Fu Y-X, Weichselbaum RR, The intersection of radiotherapy and immunotherapy: Mechanisms and clinical implications. Science Immunology 1, eaag1266 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bryant AK, Banegas MP, Martinez ME, Mell LK, Murphy JD, Trends in Radiation Therapy among Cancer Survivors in the United States, 2000–2030. Cancer Epidemiology Biomarkers & Prevention 26, 963–970 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Hou Y, Liang HL, Yu X, Liu Z, Cao X, Rao E, Huang X, Wang L, Li L, Bugno J, Fu Y, Chmura SJ, Wu W, Luo SZ, Zheng W, Arina A, Jutzy J, McCall AR, Vokes EE, Pitroda SP, Fu YX, Weichselbaum RR, Radiotherapy and immunotherapy converge on elimination of tumor-promoting erythroid progenitor cells through adaptive immunity. Science Translational Medicine 13, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arina A, Gutiontov SI, Weichselbaum RR, Radiotherapy and Immunotherapy for Cancer: From “Systemic” to “Multisite”. Clinical Cancer Research 26, 2777–2782 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weichselbaum RR, Liang H, Deng L, Fu Y-X, Radiotherapy and immunotherapy: a beneficial liaison? Nature Reviews Clinical Oncology 14, 365–379 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharyya T, Purushothaman K, Vadakke Puthiyottil SS, Bhattacharjee A, Muttah G, Immunological interactions in radiotherapy—opening a new window of opportunity. Annals of Translational Medicine 4, 51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodríguez-Ruiz ME, Vanpouille-Box C, Melero I, Formenti SC, Demaria S, Immunological Mechanisms Responsible for Radiation-Induced Abscopal Effect. Trends in Immunology 39, 644–655 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li X-D, Mauceri H, Beckett M, Darga T, Huang X, Gajewski TF, Chen ZJ, Fu Y-X, Weichselbaum Ralph R., STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843–852 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turchan WT, Pitroda SP, Weichselbaum RR, Radiotherapy and Immunotherapy Combinations in the Treatment of Patients with Metastatic Disease: Current Status and Future Focus. Clinical Cancer Research 27, 5188–5194 (2021). [DOI] [PubMed] [Google Scholar]
- 10.Turchan WT, Pitroda SP, Weichselbaum RR, Combined radio-immunotherapy: An opportunity to increase the therapeutic ratio of oligometastasis-directed radiotherapy. Neoplasia 27, 100782 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weichselbaum RR, Pitroda SP, Immunoradiotherapy goes club(bing). Nature Cancer 2, 871–872 (2021). [DOI] [PubMed] [Google Scholar]
- 12.Muroyama Y, Nirschl TR, Kochel CM, Lopez-Bujanda Z, Theodros D, Mao W, Carrera-Haro MA, Ghasemzadeh A, Marciscano AE, Velarde E, Tam AJ, Thoburn CJ, Uddin M, Meeker AK, Anders RA, Pardoll DM, Drake CG, Stereotactic Radiotherapy Increases Functionally Suppressive Regulatory T Cells in the Tumor Microenvironment. Cancer Immunology Research 5, 992–1004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kachikwu EL, Iwamoto KS, Liao Y-P, DeMarco JJ, Agazaryan N, Economou JS, McBride WH, Schaue D, Radiation Enhances Regulatory T Cell Representation. International Journal of Radiation Oncology*Biology*Physics 81, 1128–1135 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu S, Sun X, Luo J, Zhu H, Yang X, Guo Q, Song Y, Sun X, Effects of radiation on T regulatory cells in normal states and cancer: mechanisms and clinical implications. American Journal of Cancer Research 5, 3276–3285 (2015). [PMC free article] [PubMed] [Google Scholar]
- 15.Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, Zheng W, Mauceri H, Mack M, Xu M, Fu Y-X, Weichselbaum RR, Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nature Communications 8, 1736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, Dou X, Chen S, Yu X, Huang X, Zhang L, Chen Y, Wang J, Yang K, Bugno J, Pitroda S, Ding X, Piffko A, Si W, Chen C, Jiang H, Zhou B, Chmura SJ, Luo C, Liang HL, He C, Weichselbaum RR, YTHDF2 inhibition potentiates radiotherapy antitumor efficacy. Cancer Cell 41, 1294–1308.e1298 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knitz MW, Bickett TE, Darragh LB, Oweida AJ, Bhatia S, Court BV, Bhuvane S, Piper M, Gadwa J, Mueller AC, Nguyen D, Nangia V, Osborne DG, Bai X, Ferrara SE, Boss M-K, Goodspeed A, Burchill MA, Tamburini BAJ, Chan ED, Pickering CR, Clambey ET, Karam SD, Targeting resistance to radiation-immunotherapy in cold HNSCCs by modulating the Treg-dendritic cell axis. Journal for ImmunoTherapy of Cancer 9, e001955 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piper M, Van Court B, Mueller A, Watanabe S, Bickett T, Bhatia S, Darragh LB, Mayeda M, Nguyen D, Gadwa J, Knitz M, Corbo S, Morgan R, Lee J-J, Dent A, Goodman K, Messersmith W, Schulick R, Del Chiaro M, Zhu Y, Kedl RM, Lenz L, Karam SD, Targeting Treg-Expressed STAT3 Enhances NK-Mediated Surveillance of Metastasis and Improves Therapeutic Response in Pancreatic Adenocarcinoma. Clinical Cancer Research 28, 1013–1026 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oweida AJ, Darragh L, Phan A, Binder D, Bhatia S, Mueller A, Court BV, Milner D, Raben D, Woessner R, Heasley L, Nemenoff R, Clambey E, Karam SD, STAT3 Modulation of Regulatory T Cells in Response to Radiation Therapy in Head and Neck Cancer. JNCI: Journal of the National Cancer Institute 111, 1339–1349 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanaka A, Sakaguchi S, Targeting Treg cells in cancer immunotherapy. European Journal of Immunology 49, 1140–1146 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Li Q, Lu J, Li J, Zhang B, Wu Y, Ying T, Antibody-based cancer immunotherapy by targeting regulatory T cells. Frontiers in Oncology 13, 1157345 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bosteels V, Maréchal S, De Nolf C, Rennen S, Maelfait J, Tavernier SJ, Vetters J, Van De Velde E, Fayazpour F, Deswarte K, Lamoot A, Van Duyse J, Martens L, Bosteels C, Roelandt R, Emmaneel A, Van Gassen S, Boon L, Van Isterdael G, Guillas I, Vandamme N, Höglinger D, De Geest BG, Le Goff W, Saeys Y, Ravichandran KS, Lambrecht BN, Janssens S, LXR signaling controls homeostatic dendritic cell maturation. Science Immunology 8, eadd3955 (2023). [DOI] [PubMed] [Google Scholar]
- 23.Ness S, Lin S, Gordon JR, Regulatory Dendritic Cells T Cell Tolerance, and Dendritic Cell Therapy for Immunologic Disease. Frontiers in Immunology 12, 633436 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maldonado RA, Von Andrian UH, How tolerogenic dendritic cells induce regulatory T cells. Advances in Immunology 108, 111–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merad M, Sathe P, Helft J, Miller J, Mortha A, The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annual Review of Immunology 31, 563–604 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mildner A, Jung S, Development and Function of Dendritic Cell Subsets. Immunity 40, 642–656 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseño CG, Iwata A, Kretzer NM, Durai V, Murphy KM, Transcriptional Control of Dendritic Cell Development. Annual Review of Immunology 34, 93–119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, Krishnan I, Maroni G, Meyerovitz CV, Kerwin CM, Choi S, Richards WG, De Rienzo A, Tenen DG, Bueno R, Levantini E, Pittet MJ, Klein AM, Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 50, 1317–1334.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gardner A, Ruffell B, Dendritic Cells and Cancer Immunity. Trends in Immunology 37, 855–865 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, Yona S, Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nature Reviews Immunology 14, 571–578 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blair TC, Bambina S, Alice AF, Kramer GF, Medler TR, Baird JR, Broz ML, Tormoen GW, Troesch V, Crittenden MR, Gough MJ, Dendritic Cell Maturation Defines Immunological Responsiveness of Tumors to Radiation Therapy. The Journal of Immunology 204, 3416–3424 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pilones KA, Charpentier M, Garcia-Martinez E, Daviaud C, Kraynak J, Aryankalayil J, Formenti SC, Demaria S, Radiotherapy Cooperates with IL15 to Induce Antitumor Immune Responses. Cancer Immunology Research 8, 1054–1063 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, Tsui J, Ruhland MK, Kersten K, Abushawish MA, Spasic M, Giurintano JP, Chan V, Daud AI, Ha P, Ye CJ, Roberts EW, Krummel MF, Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4+ T Cell Immunity. Cell 177, 556–571.e16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bosteels C, Neyt K, Vanheerswynghels M, van Helden MJ, Sichien D, Debeuf N, De Prijck S, Bosteels V, Vandamme N, Martens L, Saeys Y, Louagie E, Lesage M, Williams DL, Tang S-C, Mayer JU, Ronchese F, Scott CL, Hammad H, Guilliams M, Lambrecht BN, Inflammatory Type 2 cDCs Acquire Features of cDC1s and Macrophages to Orchestrate Immunity to Respiratory Virus Infection. Immunity 52, 1039–1056.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duong E, Fessenden TB, Lutz E, Dinter T, Yim L, Blatt S, Bhutkar A, Wittrup KD, Spranger S, Type I interferon activates MHC class I-dressed CD11b+ conventional dendritic cells to promote protective anti-tumor CD8+ T cell immunity. Immunity 55, 308–323.e9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, Chudnovskiy A, Maskey S, Walker L, Finnigan JP, Kirkling ME, Reizis B, Ghosh S, D’Amore NR, Bhardwaj N, Rothlin CV, Wolf A, Flores R, Marron T, Rahman AH, Kenigsberg E, Brown BD, Merad M, A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580, 257–262 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, Modak M, Carotta S, Haslinger C, Kind D, Peet GW, Zhong G, Lu S, Zhu W, Mao Y, Xiao M, Bergmann M, Hu X, Kerkar SP, Vogt AB, Pflanz S, Liu K, Peng J, Ren X, Zhang Z, Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 179, 829–845.e20 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, Fu Y-X, Auh SL, The Efficacy of Radiotherapy Relies upon Induction of Type I Interferon–Dependent Innate and Adaptive Immunity. Cancer Research 71, 2488–2496 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deng L, Liang H, Fu S, Weichselbaum RR, Fu Y-X, From DNA Damage to Nucleic Acid Sensing: A Strategy to Enhance Radiation Therapy. Clinical Cancer Research 22, 20–25 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Musella M, Manic G, De Maria R, Vitale I, Sistigu A, Type-I-interferons in infection and cancer: Unanticipated dynamics with therapeutic implications. Oncoimmunology 6, e1314424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borden EC, Interferons α and β in cancer: therapeutic opportunities from new insights. Nature Reviews Drug Discovery 18, 219–234 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Trinchieri G, Type I interferon: friend or foe? Journal of Experimental Medicine 207, 2053–2063 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Einhorn S, Grander D, Why Do So Many Cancer Patients Fail to Respond to Interferon Therapy? Journal of Interferon & Cytokine Research 16, 275–281 (1996). [DOI] [PubMed] [Google Scholar]
- 44.Medrano RFV, Hunger A, Mendonça SA, Barbuto JAM, Strauss BE, Immunomodulatory and antitumor effects of type I interferons and their application in cancer therapy. Oncotarget 8, 71249–71284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salmon P, Cotonnec J-YL, GalazKa A, Abdul-Ahad A, Darragh A, Pharmacokinetics and Pharmacodynamics of Recombinant Human Interferon-β in Healthy Male Volunteers. Journal of Interferon & Cytokine Research 16, 759–764 (1996). [DOI] [PubMed] [Google Scholar]
- 46.Sleijfer S, Bannink M, Gool AR, Kruit WHJ, Stoter G, Side Effects of Interferon-α Therapy. Pharmacy World and Science 27, 423–431 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Thomas R, Weihua Z, Rethink of EGFR in Cancer With Its Kinase Independent Function on Board. Frontiers in Oncology 9, 800 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang X, Zhang X, Fu May L., Weichselbaum Ralph R., Gajewski Thomas F., Guo Y, Fu Y-X, Targeting the Tumor Microenvironment with Interferon-β Bridges Innate and Adaptive Immune Responses. Cancer Cell 25, 37–48 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei H, Cai H, Jin Y, Wang P, Zhang Q, Lin Y, Wang W, Cheng J, Zeng N, Xu T, Zhou A, Structural basis of a novel heterodimeric Fc for bispecific antibody production. Oncotarget 8, 51037–51049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin Y, Cao Y, Kim HJ, Salim A, Speed TP, Lin DM, Yang P, Yang JYH, scClassify: sample size estimation and multiscale classification of cells using single and multiple reference. Molecular Systems Biology 16, e9389 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, He Y, Wang L, Zhang Q, Kim A, Gao R, Orf J, Wang T, Sawant D, Kang J, Bhatt D, Lu D, Li C-M, Rapaport AS, Perez K, Ye Y, Wang S, Hu X, Ren X, Ouyang W, Shen Z, Egen JG, Zhang Z, Yu X, Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 181, 442–459.e29 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu Y-X, Irradiation and anti–PD-L1 treatment synergistically promote antitumor immunity in mice. The Journal of Clinical Investigation 124, 687–695 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Farren MR, Sayegh L, Ware MB, Chen H-R, Gong J, Liang Y, Krasinskas A, Maithel SK, Zaidi M, Sarmiento JM, Kooby D, Patel P, El-Rayes B, Shaib W, Lesinski GB, Immunologic alterations in the pancreatic cancer microenvironment of patients treated with neoadjuvant chemotherapy and radiotherapy. JCI Insight 5, e130362 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goldman MJ, Craft B, Hastie M, Repečka K, Mcdade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, Zhu J, Haussler D, Visualizing and interpreting cancer genomics data via the Xena platform. Nature Biotechnology 38, 675–678 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bernstein MB, Krishnan S, Hodge JW, Chang JY, Immunotherapy and stereotactic ablative radiotherapy (ISABR): a curative approach? Nature Reviews Clinical Oncology 13, 516–524 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, Beckett M, Sharma R, Chin R, Tu T, Weichselbaum RR, Fu Y-X, Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood 114, 589–595 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Palma DA, Olson R, Harrow S, Gaede S, Louie AV, Haasbeek C, Mulroy L, Lock M, Rodrigues GB, Yaremko BP, Schellenberg D, Ahmad B, Senthi S, Swaminath A, Kopek N, Liu M, Moore K, Currie S, Schlijper R, Bauman GS, Laba J, Qu XM, Warner A, Senan S, Stereotactic Ablative Radiotherapy for the Comprehensive Treatment of Oligometastatic Cancers: Long-Term Results of the SABR-COMET Phase II Randomized Trial. Journal of Clinical Oncology 38, 2830–2838 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lehrer EJ, Singh R, Wang M, Chinchilli VM, Trifiletti DM, Ost P, Siva S, Meng M-B, Tchelebi L, Zaorsky NG, Safety and Survival Rates Associated With Ablative Stereotactic Radiotherapy for Patients With Oligometastatic Cancer. JAMA Oncology 7, 92–106 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hou Y, Liang H, Rao E, Zheng W, Huang X, Deng L, Zhang Y, Yu X, Xu M, Mauceri H, Arina A, Weichselbaum RR, Fu Y-X, Non-canonical NF-κB Antagonizes STING Sensor-Mediated DNA Sensing in Radiotherapy. Immunity 49, 490–503.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, Chen X, Li X-D, Deng L, Chen ZJ, Weichselbaum RR, Fu Y-X, Dendritic Cells but Not Macrophages Sense Tumor Mitochondrial DNA for Cross-priming through Signal Regulatory Protein α Signaling. Immunity 47, 363–373.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, Saeys Y, FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry Part A 87, 636–645 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, Noelle RJ, Wang L, VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Research 74, 1933–1944 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Flies DB, Han X, Higuchi T, Zheng L, Sun J, Ye JJ, Chen L, Coinhibitory receptor PD-1H preferentially suppresses CD4+ T cell–mediated immunity. Journal of Clinical Investigation 124, 1966–1975 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Slater BT, Han X, Chen L, Xiong Y, Structural insight into T cell coinhibition by PD-1H (VISTA). Proceedings of the National Academy of Sciences 117, 1648–1657 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang X, Zhang X, Li E, Zhang G, Wang X, Tang T, Bai X, Liang T, VISTA: an immune regulatory protein checking tumor and immune cells in cancer immunotherapy. Journal of Hematology & Oncology 13, 83 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aithal KB, Kumar S, Rao BN, Udupa N, Rao SBS, Tumor Growth Inhibitory Effect of Juglone and Its Radiation Sensitizing Potential. Integrative Cancer Therapies 11, 68–80 (2012). [DOI] [PubMed] [Google Scholar]
- 67.Loschko J, Schreiber HA, Rieke GJ, Esterházy D, Meredith MM, Pedicord VA, Yao K-H, Caballero S, Pamer EG, Mucida D, Nussenzweig MC, Absence of MHC class II on cDCs results in microbial-dependent intestinal inflammation. Journal of Experimental Medicine 213, 517–534 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee W, Kim HS, Hwang SS, Lee GR, The transcription factor Batf3 inhibits the differentiation of regulatory T cells in the periphery. Experimental and Molecular Medicine 49, e393 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, Korman AJ, Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunology Research 1, 32–42 (2013). [DOI] [PubMed] [Google Scholar]
- 70.Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, Allison JP, Ribas A, Sharma P, Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3+ Regulatory T Cells (Tregs) in Human Cancers. Clinical Cancer Research 25, 1233–1238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, Peggs KS, Ravetch JV, Allison JP, Quezada SA, Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti–CTLA-4 therapy against melanoma. The Journal of Experimental Medicine 210, 1695–1710 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anz D, Rapp M, Eiber S, Koelzer VH, Thaler R, Haubner S, Knott M, Nagel S, Golic M, Wiedemann GM, Bauernfeind F, Wurzenberger C, Hornung P, Scholz Veit, C., Mayr D, Rothenfusser S, Endres S, Bourquin C, Suppression of intratumoral CCL22 by type I interferon inhibits migration of regulatory T cells and blocks cancer progression. Cancer Research 75, 4483–4493 (2015). [DOI] [PubMed] [Google Scholar]
- 73.Rapp M, Wintergerst MWM, Kunz WG, Vetter VK, Knott MML, Lisowski D, Haubner S, Moder S, Thaler R, Eiber S, Meyer B, Röhrle N, Piseddu I, Grassmann S, Layritz P, Kühnemuth B, Stutte S, Bourquin C, von Andrian UH, Endres S, Anz D, CCL22 controls immunity by promoting regulatory T cell communication with dendritic cells in lymph nodes. The Journal of Experimental Medicine 216, 1170–1181 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, Yokoi T, Chiappori A, Lee KH, De Wit M, Cho BC, Bourhaba M, Quantin X, Tokito T, Mekhail T, Planchard D, Kim Y-C, Karapetis CS, Hiret S, Ostoros G, Kubota K, Gray JE, Paz-Ares L, De Castro Carpeño J, Wadsworth C, Melillo G, Jiang H, Huang Y, Dennis PA, Özgüroğlu M, Durvalumab after Chemoradiotherapy in Stage III Non–Small-Cell Lung Cancer. New England Journal of Medicine 377, 1919–1929 (2017). [DOI] [PubMed] [Google Scholar]
- 75.Kelly RJ, Ajani JA, Kuzdzal J, Zander T, Van Cutsem E, Piessen G, Mendez G, Feliciano J, Motoyama S, Lièvre A, Uronis H, Elimova E, Grootscholten C, Geboes K, Zafar S, Snow S, Ko AH, Feeney K, Schenker M, Kocon P, Zhang J, Zhu L, Lei M, Singh P, Kondo K, Cleary JM, Moehler M, Adjuvant Nivolumab in Resected Esophageal or Gastroesophageal Junction Cancer. New England Journal of Medicine 384, 1191–1203 (2021). [DOI] [PubMed] [Google Scholar]
- 76.Turchan WT, Pitroda SP, Weichselbaum RR, Treatment of Cancer with Radio-Immunotherapy: What We Currently Know and What the Future May Hold. International Journal of Molecular Sciences 22, 9573 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liang H, Deng L, Chmura S, Burnette B, Liadis N, Darga T, Beckett MA, Lingen MW, Witt M, Weichselbaum RR, Fu Y-X, Radiation-Induced Equilibrium Is a Balance between Tumor Cell Proliferation and T Cell–Mediated Killing. The Journal of Immunology 190, 5874–5881 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, Ezoe S, Kanakura Y, Sato E, Fukumori Y, Karbach J, Jager E, Sakaguchi S, Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proceedings of the National Academy of Sciences 110, 17945–17950 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gerhard GM, Bill R, Messemaker M, Klein AM, Pittet MJ, Tumor-infiltrating dendritic cell states are conserved across solid human cancers. Journal of Experimental Medicine 218, e20200264 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, Demaria S, DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nature Communications 8, 15618 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oh SA, Wu D-C, Cheung J, Navarro A, Xiong H, Cubas R, Totpal K, Chiu H, Wu Y, Comps-Agrar L, Leader AM, Merad M, Roose-Germa M, Warming S, Yan M, Kim JM, Rutz S, Mellman I, PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nature Cancer 1, 681–691 (2020). [DOI] [PubMed] [Google Scholar]
- 82.Peng Q, Qiu X, Zhang Z, Zhang S, Zhang Y, Liang Y, Guo J, Peng H, Chen M, Fu Y-X, Tang H, PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nature Communications 11, 4835 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.