


Abstract
The lentiviral accessory protein Vpr engages an extensive network of cellular pathways to drive diverse host consequences. Of its many phenotypes, CRL4A-E3 ubiquitin ligase complex co-option, DNA damage response (DDR) engagement, and G2/M arrest are conserved and thus proposed to be functionally important. How Vpr effects these functions and whether they explain how Vpr dysregulates additional cellular pathways remain unclear. Here we leverage the ability of Vpr to deplete the nucleolar protein CCDC137 to understand how Vpr-induced DDR activation impacts nucleolar processes. We characterize CCDC137 as an indirect Vpr target whose degradation does not correlate with Vpr-induced G2/M arrest. Yet, degradation is conserved among Vpr from the pandemic HIV-1 and related SIVcpz/SIVgor, and it is triggered by genomic insults that activate a nucleolar ATR pathway in a manner similar to camptothecin. We determine that Vpr causes ATR-dependent features of nucleolar stress that correlate with CCDC137 degradation, including redistribution of nucleolar proteins, altered nucleolar morphology, and repressed ribosome biogenesis. Together, these data distinguish CCDC137 as a non-canonical Vpr target that may serve as a sensor of nucleolar disruption, and in doing so, identify a novel role for Vpr in nucleolar stress.
Graphical Abstract
Graphical Abstract.

Introduction
Lentiviral accessory proteins inhibit cellular factors and pathways that interfere with viral replication to counteract host innate immunity or augment pro-viral signaling [1]. Of the four HIV-1 accessory proteins – Vpr, Vif, Vpu, and Nef – Vpr is the only factor that has not yet been associated with a primary target(s) or function(s). Despite this, Vpr is evolutionarily conserved among all extant primate lentiviruses [2, 3] and essential for in vivo infection by HIV and SIV [4–7].
Although lentiviral Vpr has not yet been assigned a primary phenotype, the many targets and functions ascribed to it have cemented Vpr as a highly versatile protein with extensive cellular consequences. Its diverse roles include binding the host factor DCAF1 to co-opt the CRL4ADCAF1 E3 ubiquitin ligase complex and subsequently tag host proteins for degradation, arresting the cell cycle, promoting apoptosis, augmenting viral transcription, and engaging the DNA damage response (DDR) [8]. Central to these diverse functions is the host DDR, which Vpr engages through multiple, distinct mechanisms. Of the three primary DDR signaling cascades – DNA-dependent protein kinase, ataxia telangiectasia mutated (ATM), and Ataxia telangiectasia and Rad3 (ATR) kinases – Vpr activates both ATM and ATR [9–13], triggering downstream ATM-mediated NF-κB activation [13] and ATR-dependent G2/M cell cycle arrest [10], respectively. Yet, despite requiring ATR activation [10, 14], Vpr-induced cell cycle arrest cannot be explained by damage alone, as HIV-1 mutants lacking the ability to cause G2/M arrest are able to activate DDR signaling [15]. In addition to activating these kinases, Vpr further engages the host DDR by causing DNA damage, inhibiting DNA repair, and binding and degrading many proteins involved in the DDR (reviewed in [16]).
CCDC137 is a recently identified Vpr target whose degradation has been connected to Vpr-induced DNA damage, G2/M arrest, and increased viral transcription [17]. The precise cellular pathways that connect CCDC137 to these phenotypes, however, are unclear. Although CCDC137 functions are still emerging, current literature depicts CCDC137 as a multi-faceted nucleolar protein associated with diverse functions and processes. Multiple studies implicate CCDC137 overexpression in cancer progression and poor clinical outcomes [18–22], suggesting it plays an important role in regulating cellular homeostasis. In hepatocellular carcinoma (HCC), for instance, RNA binding [19] and E3 ubiquitin ligase co-option [18] by CCDC137 alter phosphoinositide 3-kinase (PI3K)/Akt signaling to promote cancer progression. In addition to its roles in tumorigenesis, CCDC137 modulates nuclear receptor signaling by sequestering the estrogen related (ERR) and retinoic acid (RAR) receptors in the nucleolus [23, 24]. The adenoviral E1A protein binds CCDC137 to disrupt its association with RAR, thus releasing RAR from the nucleolus to engage in downstream signaling [23]. Together these findings characterize CCDC137 as a versatile, though enigmatic, protein with ties to cellular proliferation. None, however, fully explain how Vpr-mediated CCDC137 degradation might contribute to Vpr-associated phenotypes, nor do they describe how CCDC137 might operate in the nucleolar compartment it inhabits.
The nucleolus is a dynamic subnuclear structure that serves as the site for the initial stages of ribosome biogenesis. Its organization is intrinsically linked to this process, encompassing three different layers representing successive steps of ribosome biogenesis, including RNA Pol I-driven ribosomal RNA (rRNA) transcription, rRNA processing, and ribosome assembly [25]. Because of the cell’s reliance on this process, the nucleolus is an essential central hub for coordinating the stress response to a variety of cellular insults [26]. Nucleolar perturbation by insults such as DNA damage, nucleotide depletion, oncogenic abnormalities, or viral infections [26, 27] are sufficient to rapidly trigger nucleolar stress that can lead to cell cycle arrest or apoptosis [28]. In addition, more recent findings demonstrate a role for the nucleolus in orchestrating cellular stress pathways that go beyond a response related to ribosome biogenesis, including DDR regulation and maintenance of genome stability [29]. In line with this, a tremendous amount of crosstalk exists between the nucleolus and DDR, as many DNA repair proteins localize to the nucleolus and, conversely, many nucleolar proteins function in the DDR [29]. Both ATM and ATR have been implicated in the nucleolar response to DDR activation depending on the cellular insult [30].
ATM and ATR are activated in the nucleolus by insults that occur both in trans, such as those in nuclear chromatin, as well as those that develop in cis, in ribosomal DNA (rDNA) [31–34]. Although the mechanisms that lead to nucleolar DDR (nDDR) activation overlap with those that trigger the global DDR, there are several distinguishing factors that render nucleolar ATR and ATM pathways unique. In response to nuclear double-stranded breaks (DSB), for instance, the MRE11-RAD50-NBS1 complex recruits and activates ATM [34, 35]; in nucleoli, NBS1 and subsequent ATM recruitment require the nucleolar protein Treacle [32]. Nucleolar ATR activation in response to rDNA damage also involves Treacle but is unique in its dependence on the ATR activator TOPBP1 [32, 36]. Upon global ATR activation, TOPBP1 and other ATR activating factors are recruited to scaffolds of single-stranded DNA (ssDNA) coated with the ssDNA binding complex RPA. In nucleoli, TOPBP1-Treacle foci act as these scaffolds, and recruit stress response factors to rDNA lesions.
Although Vpr activates and dysregulates many of the pathways implicated in nucleolar stress, a role for Vpr in nucleolar processes has yet to be defined. Here we leverage the connections between CCDC137, the nucleolus, and the DDR to test the hypothesis that Vpr modulates nucleolar pathways. We find that CCDC137 is an indirect Vpr target whose degradation is associated with, but not a direct cause or consequence of, Vpr-induced G2/M arrest. Additionally, we characterize CCDC137 as an evolutionarily important Vpr target that bridges multiple Vpr phenotypes, including a newly identified Vpr function, nucleolar stress. We further define CCDC137 as a specific component of nucleolar stress sensing that is degraded when Vpr-induced DNA damage triggers the activation of a nucleolar-specific ATR pathway. Together, these results characterize the evolutionary importance of CCDC137 depletion to Vpr function, define DDR activation as required for this depletion, and identify a role for Vpr in nucleolar processes.
Material and methods
Plasmids and generation of viruses
pcDNA3.1–3XFLAG-Vpr, pcDNA3.1–3XFLAG-Vpx, pscAAV-GFP-T2A-Vpr, pscAAV-mCherry-T2A-Vpr, and LPCX-HA-SAMHD1 plasmids were described previously [2, 37]. The pBABe-HA-ER-IPpoI plasmid was a gift from Michael Kastan (Addgene #32 565) [38]. Vpr and Vpx sequences represent the following isolates: HIV-1 Bru (H1B.FR.83.HXB2_LAI_IIIB_BRU; GenBank K03455.1), HIV-1 Q23-17 (H1.A.Q2317; GenBank AF004885.1), HIV-1 ETH220 (H1C.ET.86.ETH2220; GenBank U46016.1), HIV-1 SE6165 (H1G.SE.93.SE6165 GenBank: AF061642.1), HIV-1 SE9280 (H1J.SE.93.SE9280; GenBank AF082394.1), HIV-1 P Fr (P RBF168; GenBank GU111555.1), HIV-2 Rod9 (H2A.Rod9; GenBank M15390.1), HIV-2 7312a (H2.B.7312a; GenBank L36874.1), HIV-2 A.PT.x.ALI (H2A.PT.x.ALI; GenBank AF082339.1); HIV-2 AB.JP.04 (H2_01_AB.JP.04.NMC307_20; GenBank AB731738.1), HIV-2 B.GH.86 (H2B.GH.86.D205; GenBank X61240.1), HIV-2 ABT96 (H2G.Cl.92.ABT96; GenBank AF208027.1), SIVcpz MB897 (CPZ.CM.x.SIVcpzMB897; GenBank EF535994.1), SIVcpz EK505 (CPZ.CM.05.SIVcpzEK505; GenBank DQ373065.1), SIVcpz CAM13 (CPZ.CM.01.SIVcpzCAM13; GenBank AY169968.1), SIVgor CP684 (GOR.CM.04.SIVgorCP684com.; GenBank FJ424871), SIVsmm SL9 (SMM.SL.92.SL92B; GenBank AF334679.1), SIVsmm CFU212 (SMM.US.86.CFU212; GenBank JX860407.1), and SIVmac 239 (MAC.US.x.239; GenBank M33262). In addition, the following consensus Vpr sequences were used: HIV-1 N con [39], HIV-1 O con [39], and HIV-1 P con (MEQAPEDQGPPREPFNEWMLQTLEELKAEAVRHFPMPWLHSLGQYIYNTYGDTWEGVTAIIRILQQLIFIHYRIGCQHSRIGILTLSRRGGRHGPSRS).
LPCX-HA-CCDC137 plasmids were generated by synthesizing CCDC137 as gBlocks (IDT) that were subcloned into LPCX-SAMHD1-HA [2] using standard cloning techniques. CCDC137 protein sequences are available at Figshare: https://doi.org/10.6084/m9.figshare.26841640 and https://doi.org/10.6084/m9.figshare.26841634.
pscAAV-mCherry-T2A Vpr and pscAAV-GFP-T2A Vpr plasmids were packaged into rAAV vectors through transient transfections of Human embryonic kidney (HEK) 293 cells using polyethyleneimine as previously described [40]. Inverted terminal repeat (ITR)-specific quantitative PCR (qPCR) was used to quantify levels of DNase-resistant vector genomes as previously described [41].
Cell lines and cell culture
HEK 293T and human bone osteosarcoma epithelial (U2OS) cells were cultured as adherent cells in Dulbecco’s modified Eagle’s medium (DMEM) growth medium (high glucose, L-glutamine, no sodium pyruvate) with 10% fetal bovine serum (Peak Serum) and 1% penicillin-streptomycin (Gibco) at 37°C and 5% CO2. All cells were harvested using 0.05% trypsin-EDTA (Gibco). Transfections were performed with TransIT-LT1 (Mirus), and rAAV infections were performed by adding rAAV directly to the growth medium. For inhibitor assays, cells were incubated with 5 nm Actinomycin D (Gibco) for 5 h, 5 μM or 10 μM MLN4924 (Cell Signaling) for 8 hr, 10 μM MG132 (Sigma) for 5 hr, or 10 μM ATMi (KU-55933) (Cayman Chemicals) or ATRi (VE-821) (Cayman Chemicals) for 24 hr. For genotoxic agent assays, cells were incubated with titrating amounts of camptothecin (Cayman Chemicals), cisplatin (Tokyo Chemical Industry), etoposide (Cayman Chemicals), hydroxyurea (Sigma) or Olaparib (MedChemExpress) for 24 hr.
For CCDC137 knockdown experiments, gRNAs targeting unique sites within CCDC137 (gRNA 1: GCGACTAGATAAAGTCCGA; gRNA 2: TCAGCAAGAACCAGGCCATC) or a non-targeting gRNA control (CAGCTCATCGGTGTCCTACT) were subcloned into lentiCRISPRv2 puro (pLCV2), a gift from Brett Stringer (Addgene plasmid # 98 290; http://n2t.net/addgene:98290; RRID:Addgene_98 290) [42], using NEBridge Golden Gate Assembly (New England Biolabs). Lentivirus was produced as previously described [39]. Cells stably expressing each gRNA were selected for via 2 μg/mL puromycin (Gibco).
Flow cytometry
Approximately 6.0 × 106 cells per condition were harvested at 24 h post infection (hpi) and stained with Hoescht 33342 Ready FlowTM Reagent (Invitrogen) according to the manufacturer’s instructions for cell cycle analysis. Event counts were measured on the AttuneNxT (Thermo Fisher Scientific), and data were analyzed using FlowJo software. G1 and G2 values were obtained using the Dean-Jett-Fox Univariate model [43, 44], and G2/G1 ratios were calculated from these percentages.
Western blots and co-immunoprecipitations
Cells were lysed for western blot in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate, Universal Nuclease (Pierce), and EDTA-free protease inhibitor (Pierce) and clarified by centrifugation at 15 000 x g for 15 min. Immunoprecipitations were performed as previously described [39] using anti-HA affinity gel (Millipore Sigma) in NP-40 lysis buffer (200 mM NaCl, 50 mM Tris [pH 7.4], 0.5% NP-40, 1 mM DTT, Universal nuclease, protease inhibitor). All samples were boiled in 4X sample buffer (40% glycerol, 240 mM Tris, pH 6.8, 8% SDS, 0.5% β-mercaptoethanol, and bromophenol blue), run on 4 to 12% Bis-Tris polyacrylamide gels, and subsequently transferred onto polyvinylidene difluoride membranes (Sigma). Membranes were blocked in intercept blocking buffer (Li-COR Biosciences) and probed with the indicated primary antibodies: mouse anti-FLAG M2 1:3000 (Sigma), mouse anti-HA 1:10 000 (Invitrogen), rabbit anti-CC137 1:2000 (Atlas and Abcam), rabbit anti-VprBP 1:2000 (Proteintech), mouse anti-β-tubulin 1:5000 (Cell Signaling), rabbit anti-γH2AX 1:5000 (Cell Signaling), rabbit anti-p53 1:5000 (Proteintech), rabbit anti-UTP11 1:3000 (Antibodies Online), rabbit anti-NOP14 1:1000 (Proteintech), rabbit anti-RRP36 1:1000 (Proteintech), mouse anti-TOPBP1 1:500 (Santa Cruz Biotechnology), and rabbit anti-Ape1 1:1000 (Cell Signaling). Membranes were incubated with secondary antibodies IRDye 800CW anti-Rabbit and IRDye 680RD anti-mouse 1:20 000 (Li-COR Biosciences) and visualized using the Li-COR Odyssey M (Li-COR Biosciences).
Quantification of CCDC137 protein levels. Degradation experiments were performed as biological triplicates (n = 3), and protein levels were quantified using ImageStudio (Li-COR Biosciences). Each condition was normalized to a corresponding β-tubulin loading control. CCDC137 degradation values for a given Vpr are reported as the percentage of CCDC137 protein remaining relative to an empty vector control, averaged across three separate experiments. From these percentages, each degradation phenotype is then classified as robust (0–35% CCDC137 remaining), intermediate (36–80% CCDC137 remaining) or absent (81–100% CCDC137 remaining); these descriptions are used throughout the text to represent calculated values that fall within these ranges. Degradation heatmaps were generated based on the averaged percentages from three biological replicates (n = 3).
Quantitative Reverse transcription PCR (qRT-PCR)
Cells were harvested at 24hpi and RNA was isolated using the PureLinkTM RNA Mini Kit (Invitrogen). cDNA was generated using the SuperScript IV First-Stand Synthesis System (Invitrogen) with Oligo(dT) primers. qPCR reactions were prepared using PowerTrack SYBR Green Master Mix (Thermo Fisher Scientific) and the following primers (5′ to 3′): CCDC137 TATGAGGAGCCGCCAAGAGATG and CCTCTCCCTTTGCTTCCTTCTC, and GAPDH CAAGATCATCAGCAATGCCT and AGGGATGATGTTCTGGAGAG (IDT). All reactions were carried out on the LightCycler 480 System (Roche). To quantify mRNA levels, ΔΔCt values were calculated for each sample, and CCDC137 values were normalized to ΔΔCt values for the housekeeping gene GAPDH. Statistical data were based the averaged normalized ΔΔCt values from triplicate reactions performed for three different biological replicates.
Immunofluorescence
Cells were plated onto glass slides in 24-well plates (Greiner Bio-One) at 1.5 × 105 cells per well and allowed to expand overnight. For localization and nucleolar size assays, cells were harvested at 24 hr post-treatment. Cells were permeabilized (0.5% Triton-X 100 in PBS) for 5 min on ice prior to fixation (4% paraformaldehyde in PBS) for 20 min at 20°C and then incubated overnight in PBS at 4°C. Cells infected with the rAAV HIV-1 Q65R mutant were fixed prior to permeabilization. Coverslips were then washed three times in PBS before blocking (30% BSA, 0.1% Tween 20 in PBS) for 30 min at 20°C. Cells were incubated with the following primary antibodies diluted in blocking buffer overnight at 4°C: rabbit anti-CC137 1:200 (Abcam), mouse anti-Nucleolin 1:100 (Cell Signaling), mouse anti-UBTF 1:100 (Santa Cruz Biotechnology), mouse anti-TOPBP1 1:200 (Santa Cruz Biotechnology), mouse anti-RPA32 1:5000 (GeneTex), rabbit anti-NPM1 1:500 (Atlas), rabbit anti-Nucleolin 1:1000 (Cell Signaling) and mouse anti-FLAG M2 1:5000 (Sigma). Coverslips were then washed three times in 0.1% Tween 20 in PBS for 5min each at 20°C and incubated with Alexa Fluor-conjugated secondary antibodies (Life Technologies) for 1hr at 20°C. Nuclei were stained with Hoescht 33342 1:2000 (Invitrogen) diluted in PBS. Coverslips were mounted onto glass slides using antifade mounting media (Invitrogen).
For 5-ethynyl uridine (EU) experiments, cells were harvested at 28 hpi or 24 hr post drug treatment following a 1 hr incubation with 0.8 mM EU (Invitrogen). Coverslips were prepared according to manufacturer’s instructions for the Click-iT™ RNA Alexa Fluor™ 488 Imaging Kit (Invitrogen).
Representative images for all IF experiments were acquired on the LSM 980 using Airyscan. For quantification of Nucleophosmin 1 (NPM1) and Nucleolin (NCL) nucleoplasmic translocation, Fiji was used to quantify nucleolar or total nuclear signal measured as mean fluorescence intensity (MFI). Nucleoplasmic signal was calculated by subtracting nucleolar MFI from total nuclear MFI. Statistical analyses were based on at least 50 cells per condition. For 5-EU and nucleolar size assays, images for quantification were obtained using the Zeiss Axio Imager Z1. Fiji was used to quantify nucleolar area and EU signal, measured as MFI of total nuclei as previously described [45]. Statistical analyses were based on approximately 100–200 cells per condition. For nucleolar DDR assays, Fiji was used to quantify total vs. nucleolar number of TOPBP1 or RPA32 foci with at least 25 cells per condition. Nucleolar foci were defined by the overlap of these markers with the stable nucleolar marker NCL.
Phylogenetics and positive selection (PS) analyses
Phylogenetics, positive selection analysis, and detection of other potential genetic innovations (such as recombination, insertions/deletions and duplications) were performed using the Detections of Genetic Innovations (DGINN) pipeline, as previously described [46]. Briefly, homologous codon sequences to human CCDC137 in primates were automatically retrieved using NCBI blastn implemented in DGINN. Sequences were aligned using Prank [47] and recombination events were detected using Genetic Algorithm for Recombination Detection (GARD) [48]. Amino acid alignment at the primate level was also generated using Muscle [49]. Positive selection marks were detected in a simian codon Prank alignment of CCDC137 orthologs from 23 species using the following tests of selection in DGINN: HYPHY BUSTED and MEME, PAML codeml (M0, M1, M2, M7, M8, M8a), and Bio++ (M0, M1NS, M2NS, M7NS, M8NS, M8aNS) [50–53]. Sites under positive selection were identified using HYPHY MEME (P value < 0.05), from the codeml M2 and M8 model BEB statistics (BEB > 0.95), and Bio++ Bayesian posterior probabilities (PP) from the M2NS and M8NS models (PP > 0.95). In addition, Fast, Unconstrained Bayesian AppRoximation for Inferring Selection (FUBAR) web-based method was also used to detect site-specific positive selection (PP > 0.90) [54]. Alignments are openly available at FigShare: https://doi.org/10.6084/m9.figshare.26841640 and https://doi.org/10.6084/m9.figshare.26841634.
Protein structure prediction with AlphaFold
The AlphaFold predicted structure of the human CCDC137 protein (accession Q6PK04) was obtained from the AlphaFold database (https://alphafold.ebi.ac.uk/entry/Q6PK04) as no experimentally determined structures were available. ChimeraX was used to annotate the default CCDC137 predicted structure.
Proteomic analyses
For gene set over-representation analysis (GSOA), genes from the Greenwood et al. dataset [55] or the Johnson et al. dataset [56] were selected if they possessed a log2 fold change of at least 0.5 in either direction. GSOA was performed using the enricher function of clusterProfiler package (version 3.12.0) in R with default parameters. Significant gene ontology (GO) terms (adjusted p value < 0.05) were identified and further refined to select non-redundant terms. To select non-redundant gene sets, a GO term tree was first constructed based on distances (1-Jaccard Similarity Coefficients of shared genes in MSigDB) between the significant terms. The GO term tree was cut at h = 0.2 to identify clusters of non-redundant gene sets. For results with multiple significant terms belonging to the same cluster, the most significant term was selected (i.e. lowest adjusted p value) with the largest number of genes (i.e. to select the broadest categories). For the gene set enrichment analysis (GSEA), non-thresholded log2 fold changes from the Johnson et al. dataset were used as inputs. GO terms were not further refined. GO terms were obtained from the c5 category of Molecular Signature Database (Human MSigDB v2024.1.Hs) [57]. Cytoscape [58] was used to visualize CCDC137 interactions determined using STRING [59].
Results.
CCDC137 degradation is neither a direct cause nor consequence of G2/M arrest
To investigate the connection between CCDC137 degradation and Vpr-induced G2/M arrest, we first tested whether previously established [13] HIV-1 Vpr mutants that lack the ability to cause cell cycle arrest could still degrade CCDC137. These mutants are made in the backbone of the primary HIV-1 isolate Q23-17 (WT) and crucially separate conserved Vpr phenotypes. Consequently, they allow us to correlate CCDC137 degradation with individual Vpr functions, including G2/M arrest (H71R, S79A, R80A) and decreased DCAF1 binding (H71R) [60–62]. As a negative Vpr control, we also assayed the degradation capacity of the functionally dead Q65R mutant, which does not cause arrest, bind DCAF1, or properly localize within the nucleus [13]. To test this, we delivered either HIV-1 WT Vpr or mutants to U2OS cells via a recombinant adeno-associated virus (rAAV) vector system [15] and assayed Vpr-induced cell cycle arrest via flow cytometry (Fig. 1A and Supplementary Fig. S1A) and endogenous CCDC137 levels via quantitative western blot (Fig. 1B and C).
Figure 1.

CCDC137 degradation does not correlate with Vpr-mediated G2/M arrest. (A) Representative flow cytometry cell cycle plots of U2OS cells infected with the indicated WT or mutant Vpr and gated for Vpr (mCherry) expression at 28 h post infection (hpi). G2/G1 values were calculated from the percentages of cells in G2 and G1 in each condition based on DNA content (Hoechst fluorescence) (n = 3). (B) Representative western blot of cells harvested from the same experiment as in (A) probed for endogenous CCDC137 and 3XFLAG Vpr. (C) Quantification of CCDC137 protein levels from (B) normalized to Tubulin, and relative to the empty vector control. Data from three biological replicates are shown. (D) Heatmap of G2/G1 values from (A) and average CCDC137 levels from (B) in the presence of the indicated Vpr. Color gradients were set to empty vector values (dark blue) and either the lowest CCDC137 level or highest G2/M value (white). (E) Representative western blot of CRISPR-Cas9 edited U2OS cells stably expressing gRNAs targeting CCDC137 (KD 1 or 2) or a NTC and probed and quantified (F) as in (B and C). (G) Representative flow cytometry cell cycle plots of control or knockdown U2OS cells infected with EV or WT Vpr as in (A). (H) Heatmap of G2/G1 values from (G) and average CCDC137 levels from (E) treated with EV or WT Vpr in the indicated U2OS lines. Color gradients set as in (D). All results were analyzed by unpaired t-tests. Asterisks indicate statistical significance from empty vector control; error bars represent ± standard deviation (ns = not significant, *P 0.0332, **P
0.0332, **P 0.0021, ***P
0.0021, ***P 0.0002, ****P
0.0002, ****P 0.0001). EV = empty vector; NTC = non-targeting control; KD = knockdown.
0.0001). EV = empty vector; NTC = non-targeting control; KD = knockdown.
In the presence of HIV-1 WT Vpr, we observed a correlation between G2/M arrest and CCDC137 degradation as previously reported [17, 63]. HIV-1 WT Vpr degraded endogenous CCDC137 (50%) and resulted in potent cell cycle arrest (G2/G1 = 1.36) when compared to an empty vector control (G2/G1 = 0.46) (Fig. 1A–C). The functionally dead HIV-1 Q65R Vpr mutant failed to either cause arrest (G2/G1 = 0.42) or degrade CCDC137 (96%), as expected. When we introduced other HIV-1 Vpr mutants, however, we saw that all significantly degraded CCDC137 despite their inability to arrest cells in G2/M. The cell cycle arrest-deficient mutants HIV-1 R80A, S79A, and H71R resulted in arrest profiles similar to the empty vector control (G2/G1 = 0.41, 0.59, and 0.33, respectively) but degraded CCDC137 to levels induced by HIV-1 WT Vpr (38%, 51%, and 66%, respectively). Comparing the degradation and G2/G1 values side by side in a heatmap further highlighted a lack of correlation among the two phenotypes (Fig. 1D).
To further investigate a connection between CCDC137 depletion and G2/M arrest, we next generated stable CCDC137 knockdown (KD) cell lines and assayed their arrest profiles. Consistent with CCDC137 being essential for proliferating cell viability [17], we were able to knockdown CCDC137 to levels below those induced by HIV-1 WT Vpr using two unique gRNAs (23% for KD 1 and 21% for KD 2) (Fig. 1E and F) but could not generate full CCDC137 knockouts in the context of either gRNA pools or single-cell clones. Although expression of HIV-1 Vpr resulted in potent G2/M arrest in cells expressing a non-targeting control (NTC) gRNA (G2/G1 = 2.5) or two separate CCDC137-targeting gRNAs (G2/G1 = 2.7 for KD 1 and 2.0 for KD 2), knocking down CCDC137 in the absence of Vpr was not sufficient to trigger arrest (G2/G1 = 0.53 for NTC, 0.72 for KD 1, and 0.38 for KD 2) (Fig. 1G and Supplementary Fig. S1B).
Overall, the lack of correlation among G2/M arrest profiles and CCDC137 levels induced by either HIV-1 Vpr mutants or CCDC137-targeting gRNAs (Fig. 1H) shows that Vpr-mediated CCDC137 degradation is neither a direct cause nor consequence of cell cycle arrest.
CCDC137 degradation is not mediated by canonical Vpr engagement of the CRL4-E3 ubiquitin ligase complex
We next asked whether recruitment of the CRL4ADCAF1 E3 ubiquitin ligase complex was required for CCDC137 degradation, consistent with other canonical Vpr targets [64]. First, we assayed whether this degradation was cullin-ring ligase (CRL) dependent by co-expressing hemagglutinin (HA)-tagged human CCDC137 and 3XFLAG-tagged Vpr in the presence or absence of increasing concentrations of the neddylation inhibitor MLN4924. As a positive control, we assayed MLN4924 rescue of Vpx-mediated SAMHD1 degradation, which is also CRL4ADCAF1 dependent. MLN4924 increased CCDC137 levels in the presence of Vpr by 3.75-fold (12% to 45% increase), similar to the positive control SAMHD1 (4.5-fold, from 15% to 66%) (Fig. 2A–B). However, unlike SAMHD1, MLN4924 increased CCDC137 levels in cells lacking Vpr (1.45-fold change, from 100% to 145%), suggesting that basal CCDC137 levels may be regulated by a cullin-dependent pathway independent of Vpr. We observed similar trends when we assayed MLN4924 rescue of endogenous CCDC137 depleted by HIV-1 Vpr (Supplementary Fig. S1C and D). Moreover, Vpr-mediated depletion of both overexpressed and endogenous CCDC137 levels was also rescued by the proteosome inhibitor MG132 (Supplementary Fig. S1E–H), and CCDC137 RNA levels were not decreased in Vpr-treated samples (Fig. 2C). Together, this suggests that CCDC137 levels are regulated post-transcriptionally in a cullin-dependent manner, both dependent and independent of Vpr.
Figure 2.

CCDC137 degradation does not correlate with Vpr-mediated CRL4ADCAF1 E3 ubiquitin ligase complex recruitment. (A) MLN4924 rescue of Vpr-mediated CCDC137 degradation or Vpx-mediated SAMHD1 degradation in U2OS cells treated with MLN4924 (10 μM for SAMHD1 and 5 μM or 10 μM for CCDC137, as indicated). Western blot is representative of three separate experiments (n = 3) and probed for HA-CCDC137 or HA-SAMHD1, 3XFLAG Vpr or Vpx, and Tubulin. (B) Quantification of (A) as described for (Fig. 1C). (C) qRT-PCR of endogenous CCDC137 mRNA levels in the presence of HIV-1 WT or Q65R Vpr. (D) Co-immunoprecipitation assays against HA-SAMHD1 or HA-CCDC137 in U2OS cells treated with 10 μM MLN4924, probed for HA, 3X FLAG Vpr or Vpx, and DCAF1. All results were analyzed by unpaired t-tests. Asterisks indicate statistical significance from empty vector control; error bars represent ± standard deviation (ns = not significant, *P 0.0332, **P
0.0332, **P 0.0021, ***P
0.0021, ***P 0.0002, ****P
0.0002, ****P 0.0001). EV = empty vector.
0.0001). EV = empty vector.
To further clarify a role for CRL4ADCAF1 ubiquitin ligase complex recruitment in Vpr-mediated CCDC137 degradation, we next performed co-immunoprecipitations (co-IPs) to probe for the recruitment of DCAF1, the adaptor protein between Vpr and CRL4A-DDB1 [65], to a Vpr-CCDC137 complex. In the presence of MLN4924, we transiently co-expressed HA-tagged human CCDC137 and 3X-FLAG tagged Vpr in U2OS cells, performed IPs against HA-CCDC137, and probed for interactions with 3X-FLAG Vpr and endogenous DCAF1 (Fig. 2D). As has been previously reported, HIV-2 Rod9 Vpx robustly interacted with human SAMHD1, and DCAF1 was only recruited in the presence of HIV-2 Rod9 Vpx [66, 67]. In contrast, we only detected minimal interaction between CCDC137 and HIV-1 WT Vpr. The intensity of this interaction was no greater than that observed between CCDC137 and HIV-2 Rod9 Vpx, which does not degrade CCDC137. Moreover, low levels of DCAF1 were already bound to CCDC137 in the absence of Vpr when compared to an empty vector control, and these levels did not increase in the presence of HIV-1 WT Vpr. HIV-1 Vpr mutants that could or could not degrade CCDC137, H71R and Q65R, respectively, showed similar weak band intensities for 3XFLAG-Vpr and DCAF1 that we observed with WT Vpr. Performing the reverse-IP, we confirmed that neither HIV-1 WT nor HIV-1 Q65R Vpr interacted with CCDC137, despite the former robustly interacting with DCAF1 (Supplementary Fig. S1I). Consistent with this minimal interaction, we also did not observe co-localization among Vpr and CCDC137 via immunofluorescence (IF), despite dimmer nucleolar CCDC137 signal in cells expressing Vpr orthologs that were capable of degradation (Supplementary Fig. S2A).
To elucidate whether Vpr-mediated CCDC137 depletion might derive from altered CCDC137 dynamics within the nucleolus, we further visualized CCDC137 localization in the context of two other nucleolar proteins, nucleolin (NCL) and upstream binding transcription factor (UBF1), via IF. NCL localizes to dense fibrillar component and granular component (GC) layers of the nucleolus, reflecting its roles in rRNA processing [68], whereas UBF1 is an essential component of the RNA Polymerase I pre-initiation complex found in the fibrillar center (FC) [69]. Consistent with its localization to the FC, CCDC137 signal overlaps with that of NCL (Supplementary Fig. S2B) and borders that of UBTF (Supplementary Fig. S2C). Relative to these markers, CCDC137 appeared on the exterior of nucleoli, where it may have the potential to interact with nucleoplasmic factors. HIV-1 Vpr did not alter CCDC137 localization in the context of either protein, despite dampening CCDC137 signal. While the addition of MLN4924 rescued this loss of signal, it did not reveal CCDC137 localization in any other nuclear compartments, countering the possibility that Vpr might be mis-localizing CCDC137 prior to degradation. UBF1 signal was enhanced in the context of MLN4924, however, suggesting an additional example of a nucleolar protein whose levels may be cullin regulated.
Overall, Vpr depleted CCDC137 in the absence of a robust direct interaction, additional DCAF1 recruitment, or altered CCDC137 localization. While cellular CCDC137 levels may be regulated via a DCAF1-dependent mechanism, Vpr is not enhancing this interaction. Together, this suggests that Vpr does not degrade CCDC137 via canonical co-option of the CRL4ADCAF1 ubiquitin ligase complex.
The ability to deplete CCDC137 is evolutionarily conserved among Vpr orthologs from the HIV-1 lineage
Although we did not find evidence for a direct correlation between Vpr-mediated CCDC137 degradation and G2/M arrest, we reasoned that this degradation may still be an evolutionarily conserved, and thus important, capacity of Vpr. We first asked whether CCDC137 degradation was conserved within the broader Vpr diversity from HIV-1 and HIV-2, which both infect humans but have distinct evolutionary origins: HIV-1 originating from spillovers of SIVcpz and SIVgor, and HIV-2 from SIVsmm [70]. Using transient co-transfections of HA-CCDC137 and 3X-FLAG Vpr, we determined that four Vpr isolates from the pandemic HIV-1 group M viruses robustly degraded human CCDC137 (all < 20%), as did a consensus Vpr from group O (3.5%). A consensus Vpr from group N and two Vpr isolates from group P (one a primary sequence and one a consensus Vpr) did not significantly degrade CCDC137 (68%, and 96 and > 100%, respectively) (Fig. 3A–B). In contrast to HIV-1 Vpr, HIV-2 Vpr isolates showed intermediate or no degradation of human CCDC137, while Vpx showed no degradation (>35% CCDC137 remaining) (Supplementary Fig. S3A and B), suggesting this function is not present among HIV-2 viruses.
Figure 3.

Degradation of CCDC137 is conserved among the HIV-1/SIVcpz/SIVgor Vpr lineage. (A) Representative western blots of U2OS cells transiently transfected with human HA-CCDC137 and 3XFLAG HIV-1 Vpr isolates. Cells were harvested at 28 h post-transfection, and western blots were probed and (B) quantified as in Fig. 1C (n = 3). (C) Representative western blots and (D) quantification of chimpanzee (CPZ) HA-CCDC137 degradation by 3XFLAG SIVcpz or SIVgor Vpr isolates as described in (A) and (B) (n = 3). (E) Heatmap of HA-CCDC137 levels in the presence of Vpr/Vpx from the HIV-1 or HIV-2 lineage. The color gradient was set to the highest (dark blue) and lowest (white) CCDC137 levels measured. The corresponding cladogram represents the aligned sequences of the Vpr/Vpx orthologs used in (A–D) and SF3 (A–D) as indicated. Results were analyzed by unpaired t-tests. Asterisks indicate statistical significance from empty vector control; error bars represent ± standard deviation (ns = not significant, *P 0.0332, **P
0.0332, **P 0.0021, ***P
0.0021, ***P 0.0002, ****P
0.0002, ****P 0.0001). EV = empty vector; con. = consensus.
0.0001). EV = empty vector; con. = consensus.
We next explored whether the ability of Vpr to degrade CCDC137 was conserved among the ancestral lineages of HIV-1 and HIV-2 viruses. Within the HIV-1 lineage, we found that all Vpr isolates tested from SIV infecting chimpanzees (SIVcpz) and gorilla (SIVgor) robustly degraded chimpanzee (Pan troglodytes troglodytes) CCDC137 at each concentration tested (Fig. 3C and D). Among the HIV-2 lineage, SIV sooty mangabeys (SIVsmm) Vpr isolate SL9, which is phylogenetically distant from the other HIV-2 and SIVsmm isolates, robustly degraded CCDC137 from sooty mangabeys (Cercocebus atys). However, SIVsmm CFU212 and SIVmac239 (from macaque) Vpr had intermediate degradation phenotypes, in line with other HIV-2 isolates (Supplementary Fig. S3C and D). These results suggest that the ability to degrade CCDC137 is conserved among Vpr from HIV-1/SIVcpz/gor but not the HIV-2 lineage and may thus be a specific and important function of HIV-1 and SIVcpz/gor (Fig. 3E).
Specificity of the Vpr-mediated CCDC137 degradation is conferred by both the Vpr and CCDC137 sequence
To determine whether the degradation capacity of a given Vpr was inherent to the Vpr or dependent on the CCDC137, we challenged a panel of Vpr orthologs and isolates that could or could not degrade CCDC137 against the natural diversity of CCDC137 present in primates. We selected four Vpr orthologs based on their diverse abilities to degrade their autologous CCDC137 (Fig. 3): HIV-1 Bru (robust), HIV-2 7312a (intermediate), SIVcpz MB897 (robust), and SIVsmm CFU212 (intermediate). We assayed degradation against the breadth of CCDC137 sequence variability from humans, chimpanzees, sooty mangabeys, African Green Monkeys (AGMs) (Chlorocebus sabaeus), a prosimian (PRO, Propithecus coquereli), and a New World monkey (NWM, Callithrix jacchus). We found that HIV-1 and SIVcpz Vpr robustly degrade nearly all CCDC137 orthologs tested, including those from prosimians and NWM. However, degradation capacities of HIV-2 7312a and SIVsmm CFU212 Vpr varied, depending on the CCDC137 ortholog tested (Fig. 4A–D and Supplementary Fig. S3E and F). Together, these data suggest that degradation is an innate capacity of certain Vpr orthologs that is also influenced by unique properties of certain CCDC137 proteins (Fig. 4E). Although these phenotypes are not reminiscent of typical virus-host molecular arms-races, in which we might expect robust degradation phenotypes in the context of autologous hosts, there is some specificity conferred by both the CCDC137 (host) and Vpr (viral) sequence.
Figure 4.

Host species- and viral lineage-specific determinants influence CCDC137 degradation. (A) Representative western blot and (B) quantification of human and chimpanzee (CPZ) HA-CCDC137 degradation by a panel of 3XFLAG HIV-1 Bru, HIV-2 7312a, SIVcpz MB897, and SIVsmm CFU212 Vpr orthologs. Assays were performed as described in Fig. 3A–D (n = 3). (C) Representative western blot and quantification of (D) prosimian (PRO) and New World monkey (NWM) HA-CCDC137 degradation by the indicated Vpr (n = 3). (E) Heatmap depicting degradation of the different CCDC137 orthologs (rows) by the indicated Vpr ortholog (columns). Results were analyzed by unpaired t-tests. Asterisks indicate statistical significance from empty vector control; error bars represent ± standard deviation (ns = not significant, *P 0.0332, **P
0.0332, **P 0.0021, ***P
0.0021, ***P 0.0002, ****P
0.0002, ****P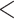 0.0001). (F) Schematic representation of the CCDC137 protein. Helices are noted as H1-5 and sites of PS (Supplementary File S1 for details and methods) are represented as large solid blue triangles. CCDC137 sequence motifs and viral protein-binding sites are represented as labeled black and red lines, respectively. (G) Residues corresponding to the proposed Vpr-binding site and marks of PS. Amino acids are labeled by property (pink = charged, teal = hydrophobic uncharged, blue = polar uncharged, and orange = special case). Grey boxes containing (-) indicate a lack of corresponding amino acid in the alignment. Sequences are organized based on an evolutionary tree (left) constructed from 28 primate CCDC137 sequences. Red asterisks indicate sequences used in (A–E). Primate icons are from Adobe Stock Images. EV = empty vector. NLS = nuclear localization sequence.
0.0001). (F) Schematic representation of the CCDC137 protein. Helices are noted as H1-5 and sites of PS (Supplementary File S1 for details and methods) are represented as large solid blue triangles. CCDC137 sequence motifs and viral protein-binding sites are represented as labeled black and red lines, respectively. (G) Residues corresponding to the proposed Vpr-binding site and marks of PS. Amino acids are labeled by property (pink = charged, teal = hydrophobic uncharged, blue = polar uncharged, and orange = special case). Grey boxes containing (-) indicate a lack of corresponding amino acid in the alignment. Sequences are organized based on an evolutionary tree (left) constructed from 28 primate CCDC137 sequences. Red asterisks indicate sequences used in (A–E). Primate icons are from Adobe Stock Images. EV = empty vector. NLS = nuclear localization sequence.
We therefore characterized the evolutionary history of CCDC137 in primates and tested whether it has experienced episodes of rapid evolution and positive selection (PS). We performed PS analyses using the Detection of Genetic INNovations (DGINN) pipeline, which performs phylogenetic and positive selection analyses from five complementary methods to increase the sensitivity and specificity in the detection of genetic innovation. Among simians, we did not find evidence of strong PS in CCDC137 (Supplementary File S1). However, performing detailed site-specific analyses, we identified five regions undergoing rapid evolution (Fig. 4F and G). Most are in predicted disordered regions, except a 269–272 region in the C-terminal helix of CCDC137 (Supplementary Fig. S3G). Furthermore, site 194 overlapped with a region linked to adenoviral E1A interaction [23], which could be reminiscent of a past evolutionary conflict. However, no signatures of positive selection or rapid evolution overlapped with nuclear localization sequences (NLS), a nuclear-hormone receptor binding motif (LXXLL), or a sequence associated with Vpr-mediated CCDC137 degradation [17]. Remarkably, the latter was entirely conserved amongst primate sequences (Fig. 4F and G). Given the functional host-specificity observed, it is thus likely that other CCDC137 determinants beyond sequence identity are also involved in CCDC137-induced degradation by Vpr.
CCDC137 depletion is a consequence of specific genomic insults
Despite our findings that CCDC137 is not a canonical target of Vpr, it is clear that Vpr orthologs from the HIV-1 lineage potently deplete this host factor. Given the absence of a direct CCDC137–Vpr interaction, we reasoned that CCDC137 depletion might result from another important HIV-1 Vpr function. Considering that CCDC137 degradation has been linked to activation of the DDR [17] and Vpr both induces DNA damage and modulates DDR signaling, we asked whether CCDC137 degradation is a consequence of DNA damage.
To test this, we investigated whether a panel of different genotoxic agents were sufficient to deplete CCDC137. Within this panel, we assayed CCDC137 levels in the context of γH2AX levels, which we used to measure the amount of global DDR activation induced by each agent. We found that titrating amounts of camptothecin (Fig. 5A and B) and etoposide (Supplementary Fig. S4A and B), which cause DNA damage by inhibiting Topoisomerase I and II, respectively, robustly depleted CCDC137. In contrast, cisplatin (Fig. 5C and D), an agent that forms intra-strand DNA crosslinks that inhibit DNA synthesis, and hydroxyurea (Supplementary Fig. S4C and D), a ribonucleotide reductase inhibitor, did not deplete CCDC137, even at the highest concentrations tested. Olaparib, a PARP1/2 inhibitor that prevents DNA damage repair and leads to an accumulation of DSBs, degraded CCDC137 only at high concentrations (Supplementary Fig. S4E and F) that may induce off-target effects. Among conditions that induced CCDC137 degradation, degradation inversely correlated with amount of DNA damage. However, CCDC137 degradation did not correlate with the absolute amount of damage present (Fig. 5E). For example, in instances where camptothecin and cisplatin both induced higher γH2AX levels than Vpr, only camptothecin resulted in CCDC137 degradation. Degradation was also independent of the ability to cause G2/M arrest (Fig. 5F) – consistent with the degradation of CCDC137 by Vpr mutants lacking this ability (Fig. 1) – or activate p53 (Fig. 5G and H). Moreover, CCDC137 depletion itself did not cause DDR activation, nor was CCDC137 required for Vpr-induced DDR activation (Supplementary Fig. S4G). Together, these results suggest that CCDC137 depletion is a consequence of specific genomic insults.
Figure 5.

CCDC137 depletion is a consequence of specific types of DNA damage. (A) Representative western blot of CCDC137 depletion following 24 h of treatment with DMSO, HIV-1 WT, or titrating amounts camptothecin performed and (B) quantified as described in Fig. 1C (n = 3). (C) Representative western blot and (D) quantification of CCDC137 and γH2AX levels in the presence of titrating amounts of cisplatin (n = 3). (E) Heatmaps indicating CCDC137 and γH2AX levels in U2OS cells treated with DMSO, HIV-1 WT Vpr, or titrating amounts of the indicated genotoxic agents. Values for CCDC137 or γH2AX in a given condition are averaged from three separate experiments (n = 3). Top row: color gradients depict the percentage of CCDC137 remaining in the empty vector control (dark blue), the lowest percent of CCDC137 measured among the represented conditions (dark pink), and CCDC137 levels in Vpr-treated cells as a midpoint (white). Bottom row: color gradients depict γH2AX induced by empty vector (dark blue), the highest γH2AX value measured among the given conditions (dark pink) and γH2AX levels induced by Vpr as a midpoint (white). (F) Representative flow cytometry plots of cell cycle profiles under the indicated conditions as described in Fig. 1A. (G) Representative western blots and (H) quantification of p53 levels following 24 h of treatment with DMSO, HIV-1 WT, camptothecin (0.5 or 5 μM), cisplatin (0.05 or 10 μM), etoposide (5 or 25 μM), hydroxyurea (0.5 μM or 10 mM), or olaparib (10 or 200 μM) (n = 3). Results were analyzed by unpaired t-tests. Asterisks indicate statistical significance from empty vector control; error bars represent ± standard deviation (ns = not significant, *P 0.0332, **P
0.0332, **P 0.0021, ***P
0.0021, ***P 0.0002, ****P
0.0002, ****P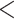 0.0001). Campto. = camptothecin; HU = hydroxyurea.
0.0001). Campto. = camptothecin; HU = hydroxyurea.
Vpr orthologs that degrade CCDC137 disrupt the nucleolar proteome
Considering that CCDC137 is a nucleolar protein, and there is an extensive crosstalk between the DDR and nucleolus, we hypothesized that CCDC137 degradation may derive from DNA damage that specifically affects nucleolar pathways. In support of nucleolar involvement, of the genotoxic agents we assayed, only those that degrade CCDC137 – camptothecin, etoposide, and olaparib – perturb the nucleolus [71–75]. Cisplatin, however, acts via nucleolar-independent DDR activation [76], in line with its inability to degrade CCDC137. To test this hypothesis, we first asked whether Vpr causes nucleolar stress.
Nucleolar stress is diagnosed based on alterations to the nucleolar proteome and changes in nucleolar morphology and function [27]. To assess the effects of Vpr on the nucleolar proteome, we leveraged two pre-existing studies that specifically assayed proteomic changes induced by Vpr [55, 56]. Johnson et al. infected Jurkat E6.1 T cells with HIV-1 ΔEnv NL4-3 (WT), HIV-1 lacking one of each accessory gene Vpr, Vpu, or Vif (ΔVpr, ΔVpu, or ΔVif, respectively), or uninfected control cells and quantified changes in the total proteome. GSEA of protein groups differentially increased or decreased among the different infection conditions revealed that the nucleolus and nucleolar processes harbored some of the most significantly decreased protein changes (Fig. 6A and Supplementary Fig. S5A). When compared to mock infection, these changes were only lost with ΔVpr infection. When compared to WT infection, only ΔVpr, but not ΔVif or ΔVpu, were statistically significant for the decrease in proteins associated with nucleolus and nucleolar processes, together showing Vpr is necessary and sufficient for alterations to the nucleolar proteome.
Figure 6.

Vpr induced nucleolar stress correlates with CCDC137 degradation. (A) GSEA of all expressed genes in [56] abundance proteomics dataset, using gene sets from the GO Cellular Component ontology. Graph legend shows normalized enrichment score computed using the enrichment and adjusted P-value values. Red arrows highlight nucleolar-associated gene sets. (B) GO terms enriched by GSOA (adjusted P-value < 0.05) of differentially expressed genes from [55] (log2FC > 0.5, Vpr expression versus empty). Top terms extracted from the GO Biological Process ontology. (C) A subnetwork extracted from STRING of CCDC137 interactions possessing a combined score greater than 0.4. (D and E) Representative western blots of nucleolar proteins found to be depleted in both abundance proteomics datasets represented in (A) and (B) in the indicated U2OS control and CCDC137 knockdown cell lines. Cells were harvested at 24hpi following treatment with EV, HIV-1 WT, or SIVsmm Vpr and the indicated protein levels were quantified in (F–I) as in Fig. 1C (n = 3). Results were analyzed by unpaired t-tests. Asterisks indicate statistical significance from the empty vector control in NTC cells; error bars represent ± standard deviation (ns = not significant, *P 0.0332, **P
0.0332, **P 0.0021, ***P
0.0021, ***P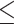 0.0002, ****P
0.0002, ****P 0.0001).
0.0001).
To understand whether Vpr-driven changes in nucleolar pathways correlated with CCDC137 degradation, we next analyzed proteomic data from Greenwood et al., in which CEM-T4 T cells were transduced with Vpr proteins that could or could not degrade CCDC137. We performed gene set overrepresentation analysis (GSOR) of differential protein abundance in each condition. In the presence of Vpr proteins that degrade CCDC137, including HIV-1 Group O, HIV-1 NL4-3, the cell cycle-deficient mutant HIV-1 S79A, and SIVcpz Vpr, the most significantly downregulated cellular pathways were nucleolar processes (Fig. 6B), particularly ribosome biogenesis. Similarly, cellular components associated with nucleolar functions harbored the most significantly depleted genes (Supplementary Fig. S5B). In contrast, among changes induced by Vpr that did not degrade CCDC137 – SIVsmm Vpr, the functionally dead mutant HIV-1 Q65R Vpr, and HIV-2 Vpx – neither nucleolar pathways nor cellular compartments harboring nucleolar processes were significantly downregulated. Further, nucleolar proteins make up the majority of proteins significantly altered by CCDC137-degrading Vpr in both the Johnson et al. and Greenwood et al. datasets (Supplementary Fig. S5C).
To determine whether CCDC137 interacted with nucleolar genes identified in the Greenwood et al. abundance dataset, we performed a functional protein association network analysis using STRING [59] (Fig. 6C). We discovered that CCDC137 interacted with multiple nucleolar proteins, including those with known roles in rRNA processing and HIV infection [77–80]. One of these proteins, nucleophomism (NPM1), is a prominent nucleolar stress sensor [81]. Together, these analyses suggest that nucleolar disruption is a specific function of Vpr, and one unique to Vpr proteins capable of CCDC137 degradation.
Given our findings that Vpr orthologs capable of depleting CCDC137 also decreased levels of other nucleolar proteins, we next sought to validate the results of our proteomic analyses and, using the CCDC137 KD cell lines (Fig. 1F), distinguish whether CCDC137 degradation was a driver of, or a response to, Vpr-mediated nucleolar disruption. We selected three nucleolar proteins that were represented in multiple nucleoli-associated GO terms in both Johnson et al. and Greenwood et al. datasets for further validation: NOP14, a stress-responsive gene involved in 40S ribosome biogenesis [82], RRP36, a component of the 90S preribosome involved in pre-rRNA processing [83], and UTP11, a subunit of the U3-containing small subunit (SSU) processome complex also involved in pre-rRNA processing [84]. We assayed degradation of each protein by HIV-1 or SIVsmm Vpr orthologs (Q23-17 and CFU212, respectively), which we focused on because of their contrasting effects on nucleolar pathways (Fig. 6B) and human CCDC137 degradation phenotypes (Fig. 4A–B). In line with the proteomic data, HIV-1 WT Vpr depleted RRP36 to levels comparable to those observed with CCDC137 in the NTC cells (46% versus 50%, respectively) (Fig. 6F–G) whereas SIVsmm Vpr only induced intermediate degradation phenotypes (74% for RRP36 versus 73% for CCDC137). Vpr-depleted RRP36 levels were also rescued by MLN4924 and MG132 (Supplementary Fig. S5D), and both etoposide and camptothecin were sufficient to induce RRP36 degradation (Supplementary Fig. S5E), further mirroring the degradation profiles of CCDC137. Although neither Vpr ortholog depleted NOP14 (Fig. 6H), both robustly degraded UTP11 (7.7% for HIV-1 WT and 5.2% for SIVsmm Vpr) (Fig. 6I), suggesting a diversity in the degradation profiles of different nucleolar proteins. CCDC137 knockdown was not sufficient to induce depletion of any of the three nucleolar proteins nor did it inhibit the ability of Vpr to induce this depletion (Fig. 6F–I), suggesting that CCDC137 is a component, but not a driver, of a larger Vpr-induced nucleolar stress response.
Vpr alters nucleolar morphology and disrupts nucleolar processes
We next determined whether Vpr triggered functional changes in nucleolar morphology and processes that are canonically associated with nucleolar stress. We first assayed redistribution of NPM1 and nucleolin (NCL) from the nucleolus to the nucleoplasm by HIV-1 WT or SIVsmm Vpr (Fig. 7A–D). As positive controls for nucleolar disruption, cells were treated with 5 nM Actinomycin D (Act. D), which selectively inhibits rRNA transcription [85], and etoposide or camptothecin (Supplementary Fig. S6A–F). Consistent with their abilities to degrade CCDC137, HIV-1 and SIVsmm Vpr led to robust and intermediate NPM1 translocation phenotypes, respectively, (Fig. 7A and B) despite inducing similar γH2AX levels (Supplementary Fig. S6G and H), suggesting NPM1 dispersal correlates with CCDC137 degradation. Moreover, this dispersal resembled the morphological changes induced by etoposide and camptothecin rather than Act. D, suggesting Vpr-induced nucleolar stress is likely not a direct derivative of RNA Pol I inhibition. While Act. D and etoposide also disrupted NCL distribution, neither the Vpr orthologs nor camptothecin altered NCL localization, further implying specificity to the nucleolar changes observed. Act. D was sufficient to induce CCDC137 depletion in a cullin-dependent manner, however, indicating that direct nucleolar insults result in CCDC137 degradation (Supplementary Fig. S6I).
Figure 7.

Vpr induces multiple signatures of nucleolar stress. (A) Representative immunofluorescence images of U2OS cells treated with the indicated conditions probed for NPM1 (magenta), 3XFLAG (Vpr) (green), and Hoechst (blue). Images were taken at 63X magnification. (B) Quantification of NPM1 translocation in (A). Nucleolar or total nuclear NPM1 MFI were measured for at least 50 cells per condition. Each dot represents one cell. (C) Representative immunofluorescence images as in (A) probed for NCL (magenta), 3XFLAG (Vpr) (green), and Hoechst (blue). (D) NCL nucleoplasmic:nucleolar translocation quantified as in (B). Results in (B) and (D) were analyzed with a one-way ANOVA with Dunnett’s multiple comparisons test. Error bars represent ± standard deviation, and asterisks indicate statistical significance from the empty vector control. (E) Representative IF images of nucleolar size experiments performed in control or CCDC137 knockdown U2OS cell lines probed for nucleolin (NCL) as described in (C). Images were taken at 63X. (F) Quantification of nucleolar size experiments performed as in (E) in the indicated cell lines. Nucleolar area was measured for approximately 100 cells per condition. Each dot represents one cell. Representative quantification of two independent experiments (n = 2). (G) Representative IF images of control or CCDC137 knockdown U2OS cells expressing HIV-1 WT Vpr or empty vector and stained for EU. Images were taken at 63X. (H) Quantification of total MFI in EU pulsed cells of the indicated conditions. MFI of EU signal was measured for approximately 100 cells per condition. Representative quantification of two independent experiments (n = 2). Results in (F) and (H) were analyzed with a one-way ANOVA with Šidák’s multiple comparison test. Error bars represent ± standard deviation, and asterisks indicate statistical significance from empty vector control or NTC cells subject to the same treatment condition; ns = not significant, *P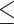 0.0332, **P
0.0332, **P 0.0021, ***P
0.0021, ***P 0.0002, ****P
0.0002, ****P 0.0001.
0.0001.
In addition to nucleolar protein redistribution, we also looked at nucleolar size, which is indicative of nucleolar disruptions [27], including those caused by viral infections [26]. We measured nucleolar area among the same conditions as in Fig. 7A–D in cells targeted with NTC or CCDC137 gRNAs (Fig. 7E and F). As expected, Act. D reduced nucleolar size, consistent with inhibited RNA Pol I activity. While HIV-1 Vpr significantly increased nucleolar size, we also observed this phenotype for SIVsmm Vpr, suggesting that augmented nucleolar area does not correlate with CCDC137 depletion. In line with this, CCDC137 knockdown was not sufficient to increase nucleolar size.
Finally, we assayed whether Vpr disrupted ribosome biogenesis and whether this correlated with CCDC137 degradation. To determine this, we assayed changes in rRNA transcription, the rate limiting step of ribosome biogenesis [86], by 5-ethynyl uridine (EU) pulse-labeling. As this step can represent 80% of cellular transcription, general changes in EU signal are indicative of changes in nascent rRNA transcription [45]. Reflecting the findings from our nucleolar size assays, we found that HIV-1 and SIVsmm Vpr both significantly decreased EU signal, suggestive of decreased nascent rRNA transcription and thus inhibition of ribosome biogenesis (Fig. 7G and H). Depletion of CCDC137 had no effect on nascent rRNA transcription, however, further suggesting that CCDC137 degradation does not directly trigger a nucleolar stress response but may be an important component of one.
Overall, these data suggest that Vpr triggers multiple nucleolar stress phenotypes, including NPM1 translocation – the most prominent hallmark of nucleolar stress [81] – increased nucleolar area, and repression of nascent rRNA transcription.
Vpr induces CCDC137 depletion through ATR-dependent nucleolar stress
Thus far, our results suggest that specific types of DNA damage are sufficient to disrupt the nucleolus and deplete CCDC137. Consequently, we sought to clarify the pathways connecting these processes. Central to the nucleolar-DDR crosstalk are the DDR signaling kinases ATM and ATR, which can be activated in response to both direct and indirect nucleolar insults [34, 87]. To determine whether activation of either kinase was required for CCDC137 depletion, we asked whether Vpr-mediated CCDC137 degradation was rescued by inhibition of ATM via KU-55933 (ATMi) or ATR via VE-821 (ATRi). While ATMi had no effect on CCDC137 degradation, ATRi fully rescued CCDC137 degradation induced by both HIV-1 and HIV-2 Vpr (Fig. 8A and B). This rescue was only observed for Vpr, given that neither inhibitor rescued CCDC137 degradation induced by camptothecin or etoposide (Supplementary Fig. S7A). Overall, these results demonstrate that CCDC137 degradation requires Vpr-induced ATR activation.
Figure 8.

Vpr induces ATR-dependent nucleolar stress thereby depleting CCDC137. (A) Representative western blots of CCDC137 levels in the presence of the indicated Vpr orthologs and DMSO, 10 μM ATMi, or 10 μM ATRi following a 24 h incubation. Western blots were probed and (B) quantified as in Fig. 1 (n = 3). Unpaired t-tests, error bars represent ± standard deviation, and asterisks indicate statistical significance from empty vector control; ns = not significant, *P 0.0332, **P
0.0332, **P 0.0021, ***P
0.0021, ***P 0.0002, ****P
0.0002, ****P 0.0001. (C) Representative IF images of nucleolar size experiments probed for nucleolin (NCL) as described in Fig. 7 (C). Images were taken at 100X. (D and E) Quantification of nucleolar size experiments performed as in (C) in ATRi (D) or ATMi (E) treated cells. Nucleolar area was measured for approximately 100 cells per condition. Representative quantification of two independent experiments (n = 2). (F) Representative IF images of U2OS cells expressing HIV-1 WT Vpr or empty vector and stained for EU. Images were taken at 63X. (G-H) Quantification of total MFI in EU pulsed cells of the indicated conditions. MFI of EU signal was measured for approximately 100 cells per condition. Representative quantification of two independent experiments (n = 2). Results in (D and E) and (G and H) were analyzed with a one-way ANOVA with Šidák’s multiple comparison test. Error bars represent ± standard deviation, and asterisks indicate statistical significance from empty vector control or cells subject to the same treatment conditions without inhibitor as indicated; ns = not significant, *P
0.0001. (C) Representative IF images of nucleolar size experiments probed for nucleolin (NCL) as described in Fig. 7 (C). Images were taken at 100X. (D and E) Quantification of nucleolar size experiments performed as in (C) in ATRi (D) or ATMi (E) treated cells. Nucleolar area was measured for approximately 100 cells per condition. Representative quantification of two independent experiments (n = 2). (F) Representative IF images of U2OS cells expressing HIV-1 WT Vpr or empty vector and stained for EU. Images were taken at 63X. (G-H) Quantification of total MFI in EU pulsed cells of the indicated conditions. MFI of EU signal was measured for approximately 100 cells per condition. Representative quantification of two independent experiments (n = 2). Results in (D and E) and (G and H) were analyzed with a one-way ANOVA with Šidák’s multiple comparison test. Error bars represent ± standard deviation, and asterisks indicate statistical significance from empty vector control or cells subject to the same treatment conditions without inhibitor as indicated; ns = not significant, *P 0.0332, **P
0.0332, **P 0.0021, ***P
0.0021, ***P 0.0002, ****P
0.0002, ****P 0.0001.
0.0001.
To specifically determine if HIV-1 Vpr induces a nucleolar ATR response that correlates with CCDC137 depletion, we assayed the number of RPA32 and TOPBP1 foci found in the nucleoli of cells treated with EV, HIV-1 Vpr, or SIVsmm Vpr. As a positive control for rDNA damage, we also measured foci induced by overexpression of I-Ppo1, an endonuclease that cleaves a recognition site within 28S rDNA and a few other locations in the genome [32, 38, 88]. We found that both HIV-1 Vpr and I-Ppo1 significantly induced nuclear and nucleolar foci of both RPA32 and TOPBP1 (Supplementary Fig. S7B–E), suggesting the activation of ATR pathways in trans and in cis. We further assayed whether Vpr activated a nucleolar ATR response by augmenting total protein levels of APE1 – a multifunctional protein whose overexpression induces nucleolar ATR activation and represses rRNA transcription in unperturbed conditions [89] – but found that Vpr did not alter total APE1 protein levels (Supplementary Fig. S7F and G). Taken together, these results suggest that HIV-1 Vpr activates multiple nucleolar markers of ATR activation.
Given that ATR activation is necessary but not sufficient for CCDC137 degradation, we hypothesized that this degradation is due to the activation of an ATR-dependent nucleolar response to DNA damage. To test this hypothesis, we asked if Vpr-induced nucleolar stress was also dependent on ATR. We first assayed nucleolar size using the same Vpr and genotoxic agent panel as in Fig. 6A and B and Supplementary Fig. S6A and B. As expected, we detected larger nucleoli among all experimental conditions apart from Act. D (Fig. 8C–E and Supplementary Fig. S8A and B). Consistent with CCDC137 degradation, ATRi rescued the altered nucleolar sizes induced by both HIV-1 and HIV-2 Vpr, but not SIVsmm Vpr. Instead, the increased nucleolar size induced by SIVsmm CFU212 was fully rescued by ATMi, suggesting that Vpr orthologs that degrade CCDC137 do so by triggering a nucleolar ATR pathway. We also found that camptothecin augmented nucleolar size in an ATR dependent manner, whereas etoposide induced changes were ATM dependent (Supplementary Fig. S8A and B). This increased nucleolar size was not solely the result of enlarged nuclei, as nucleolar number also decreased, indicative of nucleolar fusion (Supplementary Fig. S8D). Moreover, it was not a consequence of G2/M arrest, as it was observed for SIVsmm CFU212 Vpr (Fig. 8D and E), which does not induce arrest (Supplementary Fig. S8D).
We next asked whether the Vpr-mediated repression of rRNA transcription could be similarly rescued by ATR and ATM inhibition. Reflecting the nucleolar size assays, all Vpr orthologs decreased rRNA transcription; repression caused by HIV-1 and HIV-2 Vpr was ATR dependent whereas repression caused by SIVsmm CFU212 was ATM dependent (Fig. 8F–H). Similarly, camptothecin and etoposide decreased EU signal, and these changes were ATR and ATM dependent, respectively (Supplementary Fig. S8E and F).
Together, these data show that Vpr orthologs that degrade CCDC137 cause nucleolar stress by modulating the nucleolar proteome, altering NPM1 localization and nucleolar size, and decreasing ribosome biogenesis. Moreover, they suggest that HIV Vpr degrades CCDC137 through the activation of a nucleolar-specific ATR pathway, suggesting CCDC137 plays a central role in nucleolar homeostasis.
Discussion
Vpr-mediated CCDC137 depletion represents a powerful example of how indirect host targets can reveal novel lentiviral accessory protein functions and help to define novel roles for cellular proteins. In this study, we establish CCDC137 as a non-canonical, but evolutionarily important, Vpr target that is part of a broader cellular response to nucleolar stress. By leveraging a set of HIV-1 Vpr mutants and genotoxic agents that do not cause G2/M arrest, as well as CRISPR-Cas9-mediated CCDC137 depletion, we show that CCDC137 degradation is neither a direct cause or consequence of Vpr-induced cell cycle arrest. Further, we show that degradation is not mediated through canonical Vpr recruitment of the CRL4ADCAF1 ubiquitin ligase complex, and that Vpr and CCDC137 neither directly interact nor occupy the same nuclear compartments. By assaying degradation among Vpr and CCDC137 orthologs representative of the broader HIV and SIV diversity, we show that CCDC137 depletion is conserved among the HIV-1 but not HIV-2 lineage, suggesting it may constitute, or be a component of, an important function of Vpr from the pandemic HIV-1 lineage. CCDC137 depletion is also species specific and CCDC137 harbors signatures of rapid evolution, consistent with a possible evolutionary arms-race. In defining how Vpr leads to degradation of CCDC137, we show that CCDC137 is depleted by Vpr orthologs and genotoxic agents that activate an ATR-mediated nucleolar stress response. We characterize CCDC137 depletion as one component of this nucleolar stress response and show that it occurs alongside other Vpr-induced nucleolar insults, including (1) modulated nucleolar pathways, (2) altered nucleolar morphology, (3) nucleolar DNA damage, and (4) downregulated rRNA transcription. When nucleolar stress is not accompanied by CCDC137 degradation, as in the case of SIVsmm Vpr, it is ATM dependent. Together, we identify a current model where Vpr activates a nucleolar-specific ATR signaling pathway that triggers nucleolar stress and, with it, CCDC137 degradation.
Nucleolar stress may bridge Vpr-induced DNA damage and G2/M arrest. Here, we clarify that CCDC137 depletion is neither a direct cause nor consequence of cell cycle arrest. Yet, there is an apparent, albeit complex, relationship between these two phenotypes, and the temporal order of CCDC137 degradation, arrest, and nucleolar stress remains unresolved. Our results favor a process in which CCDC137 depletion precedes arrest: Vpr causes ATR-dependent nucleolar stress, of which CCDC137 degradation is a component, and this eventually triggers G2/M arrest. Given that CCDC137 depletion is not sufficient to trigger arrest, one possibility is that Vpr-induced nucleolar insults need to be severe enough to stall cell cycle progression. In support of this, the nucleolar stress response can activate an ATR-Chk1-mediated G2 arrest once a certain threshold of stress has been reached [90]. The signatures of nucleolar stress we observe are not solely consequences of arrest, given that we see NPM1 redistribution and decreased rRNA transcription among conditions with varying arrest phenotypes. Moreover, through bioinformatic analyses, we also determine that downregulation of ribosome biogenesis and other nucleolar pathways is a specific cellular consequence of Vpr but not Vif, despite both proteins causing similar cell cycle arrest [91]. In contrast to our model, others have shown that CCDC137 degradation can be caused by cell cycle arrest [63], though this may be due to the intertwined nature of arrest and nucleolar disruption, as ribosome biogenesis both regulates and is regulated by cell cycle progression. Such sensitivity of the nucleolus to disruptions may also explain why some Vpr orthologs are able to degrade only certain CCDC137 proteins in the same system. Overall, future work will be needed to establish a clearer temporal order of CCDC137 degradation, nucleolar disruption, and arrest.
By comparing the effects of Vpr and different genotoxic agents on CCDC137 levels and nucleolar integrity, our data also provide insight into how Vpr engages the DDR. Previous studies from our lab and others have shown that Vpr causes DNA damage and simultaneously activates and represses DDR pathways [16]. Here, we have demonstrated that Vpr activates a nucleolar-specific ATR pathway that is distinct from ATR triggered by general DNA damage, as all of the genotoxic agents we tested activate classical ATR [79, 80, 82], but few disrupt the nucleolus. Yet, it remains to be seen whether activation of this pathway demonstrates Vpr-mediated ATR dysregulation or represents one of numerous ATR signaling branches engaged by Vpr. Our data show that Vpr activates markers of ATR activation in rDNA, though the precise locations and nature of these foci, as well as a causal link to other ATR-dependent nucleolar stress phenotypes, will require further investigation. Moreover, it is also possible that the nucleolar ATR pathway is not activated by DNA damage but by independent nucleolar insults. What these insults may be and how they could differentially activate ATR remains to be identified. We further identify multiple phenotypes shared by HIV-1 Vpr and the Topo I inhibitor camptothecin, including robust CCDC137 degradation, and activation of ATR-dependent changes in nucleolar size and rRNA transcription. Past work has shown that Vpr induces double-stranded DNA breaks by structurally altering DNA in a manner that is Topo I and ATR dependent [92], supporting a mechanism in which Vpr might inhibit Topo I activity to activate ATR signaling. In addition, camptothecin has immediate effects on rRNA processing [71], corroborating a role for CCDC137 in this pathway, though it remains unclear whether which, if either, camptothecin mechanism Vpr phenocopies.
CCDC137 may act as a sensor of nucleolar disruption with implications for cancer progression. CCDC137 has been classified as an oncogenic RNA binding protein, but its cellular functions and precise roles in cancer progression are only beginning to be understood. Our results reveal that CCDC137 is depleted as a consequence of ATR-dependent nucleolar stress. Depletion of CCDC137 itself, however, does not result in cell cycle arrest, DNA damage, or nucleolar stress. Identifying the exact nucleolar pathways in which CCDC137 participates is the next outstanding question. Considering that the nucleolus is a cellular stress sensor, it is possible that CCDC137 degradation, like NPM1 redistribution, represents an early step in this stress response. However, it is also possible CCDC137 degradation is a consequence of specific nucleolar pathways being disrupted. Our proteomic and STRING network analyses suggest that CCDC137 may be involved in rRNA processing, the inhibition of which has been identified as a stress-dependent regulatory event in the maintenance of nucleolar integrity [93]. In line with this, other nucleolar proteins depleted by Vpr – RRP36 and UTP11 – are involved in pre-rRNA processing. This would support a model in which CCDC137 acts as a nucleolar stress sensor, is depleted in response to specific ATR-dependent stress signaling, and contributes to a pause in rRNA processing that triggers the eventual downregulation of other ribosome biogenesis pathways. Given that unprocessed rRNA is stored in the nucleolus [93], this may explain why we see increased nucleolar size with Vpr and other agents that degrade CCDC137. This storage mechanism allows for the resolution of nucleolar stress and eventual return to normal function; if the nucleolus cannot coordinate this pause in rRNA processing with other steps of ribosome biogenesis, however, it can enter a state of irreversible stress that leads to cell cycle arrest and death [94]. It is this disconnect in the ability to resume versus not recover homeostasis that may explain why some Vpr proteins are able to degrade CCDC137 without further downstream consequences, such as cell cycle arrest. Such a disconnect would also have important implications in cancer. For instance, if CCDC137 is acting as a sensor of nucleolar stress that helps dampen nucleolar processes when depleted, it is plausible that CCDC137 overexpression would prevent this ATR-dependent nucleolar inhibition. In line with this, ribosome biogenesis is hyperactivated in cancer and has been causally associated with malignant transformation and cancer progression [95]. Although there is much exploration to be done in eliciting the exact pathways involved, this study supports that CCDC137 may be a potential therapeutic target whose depletion may slow oncogenesis. Moreover, it clarifies the DNA damage pathways that influence the nucleolus and recognizes various genotoxic agents that may be used to target CCDC137 and impede nucleolar processes.
Vpr is one of many diverse viral proteins that may hijack the nucleolus to enhance viral replication. Finally, our findings raise the question of how Vpr-induced nucleolar stress might favor viral replication. Viral disruption of the nucleolus is a broad phenomenon, and numerous proteins from both plant and animal viruses localize to the nucleolus, redistribute nucleolar proteins for use in viral replication, and either enhance or inhibit ribosome biogenesis [27, 96]. For instance, in herpes simplex virus infection, nucleolar proteins localize to viral replication centers and co-localize with the viral ICP8 protein to enhance viral replication [97]. Similarly, the infectious bronchitis coronavirus (IBV) N protein localizes to the nucleolus [98], and IBV results in nucleolar enlargement similar to that seen with Vpr [99].
In the context of HIV, the nucleolar localizations of both trans-activating proteins Tat and Rev are crucial to HIV-1 replication [100–102]. Within this subnuclear compartment, Tat impairs rRNA processing [103] and remodels the nucleolar proteome [104], and Rev facilitates the export of un- and partially spliced viral RNA [105]. How the effects of Vpr on the nucleolus coordinate with these functions is unknown, though previous studies connecting Vpr to translational impairment may provide insight into such an interplay. Vpr is reported to repress cellular translation via a mechanism that does not impede the translation of viral mRNAs [106], and in doing so may ready nucleolar processes for optimal Tat and Rev exploitation. In addition, NPM1 mediates the nucleolar retention of Tat and prevents its association with the super elongation complex [107], and it is conceivable that Vpr-induced NPM1 redistribution may increase Tat nucleoplasm localization to enhance transcriptional elongation. This is consistent with the proposed role of CCDC137 depletion in HIV-1 transcription [17], which may occur as part of the stress response that releases Tat. Future work will be necessary to dissect how Vpr-induced nucleolar stress influences other HIV processes and impacts viral replication.
While there is still much to understand about the roles of CCDC137, Vpr, and the nucleolus in viral replication and cellular homeostasis, our data begin to unravel these complex connections. Moreover, we have characterized CCDC137 as an early sensor of nucleolar insults, and, in doing so, have opened new avenues for discovering further CCDC137 functions and pathways involved in the nucleolar stress response.
Supplementary Material
Acknowledgements
We are grateful to Dr. Jefferey Long and Dr. Steve Jacobsen for use of the LSM980 and Dr. Peter Bradley for use of the Zeiss Axio Imager Z1. We thank Dr. Steven Bensinger for use of the LightCycler 480 system. We thank the James B. Pendleton Charitable Trust for providing the LiCOR Odyssey M. We thank Dr. Randilea Nichols Doyle and Vivian Yang for their comments on the manuscript. We thank Andrea Cimarelli and LP2L members (CIRI) for support and discussion. We thank Laurent Guéguen (LBBE) for continuous support maintaining the DGINN pipeline. We thank all the contributors of the publicly available bioinformatic programs and genomic sequences. The graphical abstract was created in BioRender. Nisson, K. (2024) BioRender.com/o62m337.
Author contributions: K.A.N.: Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Validation, Visualization, Writing – original draft, Writing – review & editing. R.S.P.: Formal analysis, Investigation, Writing – review & editing. L.E. : Conceptualization, Data curation, Formal analysis, Investigation, Resources, Writing – review & editing. Y.D. – Formal analysis, Investigation. M.B. – Formal analysis, Resources, Supervision. O.I.F.: Conceptualization, Funding acquisition, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Contributor Information
Karly A Nisson, Molecular Biology Institute, University of California, Los Angeles, CA, 90095, United States.
Rishi S Patel, Department of Microbiology, Immunology, and Molecular Genetics, University of California, Los Angeles, CA, 90095, United States.
Yennifer Delgado, Molecular Biology Institute, University of California, Los Angeles, CA, 90095, United States.
Mehdi Bouhaddou, Molecular Biology Institute, University of California, Los Angeles, CA, 90095, United States; Department of Microbiology, Immunology, and Molecular Genetics, University of California, Los Angeles, CA, 90095, United States; Institute for Quantitative and Computational Biosciences, University of California, Los Angeles, CA, 90095, United States.
Lucie Etienne, Centre International de Recherche en Infectiologie (CIRI), Inserm U1111, UCBL1, CNRS UMR5308, ENS de Lyon, Université de Lyon, Lyon, 69007, France.
Oliver I Fregoso, Molecular Biology Institute, University of California, Los Angeles, CA, 90095, United States; Department of Microbiology, Immunology, and Molecular Genetics, University of California, Los Angeles, CA, 90095, United States; Department of Molecular, Cell & Developmental Biology, University of California, Santa Cruz, CA, 95064, United States.
Supplementary data
Supplementary data is available at NAR online.
Conflict of interest
None declared.
Funding
This work was supported by the National Institutes of Health [R01AI147837 to O.I.F.; T32AI007323 to K.A.N. and Y.D.]; a HIV Accessory and Regulatory Complexes (HARC) Collaborative Development Award [U54AI170792 to O.I.F. and M.B.]; a UCLA-CDU Center for AIDS Research (CFAR) Pilot Grant [to O.I.F. and M.B.]; a W.M. Keck Foundation Award [to O.I.F.]; the French Research Agency on HIV and Emerging Infectious Diseases ANRS/MIE [#ECTZ19143 to L.E.]; L.E. is further supported by the CNRS. Funding to pay the Open Access publication charges for this article was provided by the National Institutes of Health.
Data availability
The CCDC137 alignment data underlying this article are available in FigShare at https://figshare.com/ and can be accessed with https://doi.org/10.6084/m9.figshare.26841640 and https://doi.org/10.6084/m9.figshare.26841634.
References
- 1. Malim MH, Emerman M HIV-1 accessory proteins–ensuring viral survival in a hostile environment. Cell Host Microbe. 2008; 3:388–98. 10.1016/j.chom.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 2. Lim ES, Fregoso OI, McCoy CO et al. The ability of primate lentiviruses to degrade the monocyte restriction factor SAMHD1 preceded the birth of the viral accessory protein vpx. Cell Host Microbe. 2012; 11:194–204. 10.1016/j.chom.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tristem M, Purvis A, Quicke DL Complex evolutionary history of primate lentiviral vpr genes. Virology. 1998; 240:232–7. 10.1006/viro.1997.8929. [DOI] [PubMed] [Google Scholar]
- 4. Campbell BJ, Hirsch VM Vpr of simian immunodeficiency virus of African green monkeys is required for replication in macaque macrophages and lymphocytes. J Virol. 1997; 71:5593–602. 10.1128/jvi.71.7.5593-5602.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lang SM, Weeger M, Stahl-Hennig C et al. Importance of vpr for infection of rhesus monkeys with simian immunodeficiency virus. J Virol. 1993; 67:902–12. 10.1128/jvi.67.2.902-912.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Somasundaran M, Sharkey M, Brichacek B et al. Evidence for a cytopathogenicity determinant in HIV-1 Vpr. Proc Natl Acad Sci USA. 2002; 99:9503–8. 10.1073/pnas.142313699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beaumont T, van Nuenen A, Broersen S et al. Reversal of human immunodeficiency virus type 1 IIIB to a neutralization-resistant phenotype in an accidentally infected laboratory worker with a progressive clinical course. J Virol. 2001; 75:2246–52. 10.1128/JVI.75.5.2246-2252.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Andersen JL, Planelles V The role of Vpr in HIV-1 pathogenesis. Curr HIV Res. 2005; 3:43–51. 10.2174/1570162052772988. [DOI] [PubMed] [Google Scholar]
- 9. Lai M, Zimmerman ES, Planelles V et al. Activation of the ATR pathway by human immunodeficiency virus type 1 vpr involves its direct binding to chromatin in vivo. J Virol. 2005; 79:15443–51. 10.1128/JVI.79.24.15443-15451.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roshal M, Kim B, Zhu Y et al. Activation of the ATR-mediated DNA damage response by the HIV-1 viral protein R. J Biol Chem. 2003; 278:25879–86. 10.1074/jbc.M303948200. [DOI] [PubMed] [Google Scholar]
- 11. Nakai-Murakami C, Shimura M, Kinomoto M et al. HIV-1 Vpr induces ATM-dependent cellular signal with enhanced homologous recombination. Oncogene. 2007; 26:477–86. 10.1038/sj.onc.1209831. [DOI] [PubMed] [Google Scholar]
- 12. Zimmerman ES, Sherman MP, Blackett JL et al. Human immunodeficiency virus type 1 vpr induces DNA replication stress in vitro and in vivo. J Virol. 2006; 80:10407–18. 10.1128/JVI.01212-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sandoval C, Nisson K, Fregoso OI HIV-1 Vpr-induced DNA damage activates NF-κB through ATM-NEMO independent of cell cycle arrest. mBio. 2024; 15:e0024024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li G, Park HU, Liang D et al. Cell cycle G2/M arrest through an S phase-dependent mechanism by HIV-1 viral protein R. Retrovirology. 2010; 7:59. 10.1186/1742-4690-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li D, Lopez A, Sandoval C et al. HIV Vpr modulates the host DNA damage response at two independent steps to damage DNA and repress double-strand DNA break repair. mBio. 2020; 11:e00940-20. 10.1128/mBio.00940-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lopez A, Nichols Doyle R, Sandoval C et al. Viral modulation of the DNA damage response and innate immunity: two sides of the same coin. J Mol Biol. 2022; 434:167327. 10.1016/j.jmb.2021.167327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang F, Bieniasz PD HIV-1 Vpr induces cell cycle arrest and enhances viral gene expression by depleting CCDC137. eLife. 2020; 9:e55806. 10.7554/eLife.55806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu L, Liu Q, Liu H et al. Disrupting CCDC137-mediated LZTS2 and β-TrCP interaction in the nucleus inhibits hepatocellular carcinoma development via β-catenin and AKT. Cell Death Differ. 2025; 32:134–48. 10.1038/s41418-024-01328-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tao S, Xie S-J, Diao L-T et al. RNA-binding protein CCDC137 activates AKT signaling and promotes hepatocellular carcinoma through a novel non-canonical role of DGCR8 in mRNA localization. J Exp Clin Cancer Res. 2023; 42:134–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bai L, Yang Z-X, Liu J-S et al. Prognostic significance of CCDC137 expression and its association with immune infiltration in hepatocellular carcinoma. Dis Markers. 2022; 2022:5638675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dai W, Wu J, Peng X et al. CDK12 orchestrates super-enhancer-associated CCDC137 transcription to direct hepatic metastasis in colorectal cancer. Clin Transl Med. 2022; 12:e1087. 10.1002/ctm2.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo L, Li B, Lu Z et al. CCDC137 is a prognostic biomarker and correlates with immunosuppressive tumor microenvironment based on pan-cancer analysis. Front Mol Biosci. 2021; 8:674863. 10.3389/fmolb.2021.674863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Um S-J, Youn HS, Kim E-J De-repression of RaRF-mediated RAR repression by adenovirus E1A in the nucleolus. Biochem Biophys Res Commun. 2014; 444:605–10. 10.1016/j.bbrc.2014.01.105. [DOI] [PubMed] [Google Scholar]
- 24. Um S-J, Youn H, Kim E-J Negative regulation of errα by a novel nucleolar protein. Biochem Biophys Res Commun. 2012; 418:290–5. 10.1016/j.bbrc.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 25. Boisvert F-M, van Koningsbruggen S, Navascués J et al. The multifunctional nucleolus. Nat Rev Mol Cell Biol. 2007; 8:574–85. 10.1038/nrm2184. [DOI] [PubMed] [Google Scholar]
- 26. Boulon S, Westman BJ, Hutten S et al. The nucleolus under stress. Mol Cell. 2010; 40:216–27. 10.1016/j.molcel.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matthews D, Emmott E, Hiscox J Viruses and the nucleolus. Nucleolus. 2011; 15:321–45. 10.1007/978-1-4614-0514-6. [DOI] [Google Scholar]
- 28. Suzuki A, Kogo R, Kawahara K et al. A new PICTure of nucleolar stress. Cancer Sci. 2012; 103:632–7. 10.1111/j.1349-7006.2012.02219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ogawa LM, Baserga SJ Crosstalk between the nucleolus and the DNA damage response. Mol BioSyst. 2017; 13:443–55. 10.1039/C6MB00740F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lindström MS, Jurada D, Bursac S et al. Nucleolus as an emerging hub in maintenance of genome stability and cancer pathogenesis. Oncogene. 2018; 37:2351–66. 10.1038/s41388-017-0121-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kruhlak M, Crouch EE, Orlov M et al. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature. 2007; 447:730–4. 10.1038/nature05842. [DOI] [PubMed] [Google Scholar]
- 32. Mooser C, Symeonidou I-E, Leimbacher P-A et al. Treacle controls the nucleolar response to rDNA breaks via TOPBP1 recruitment and ATR activation. Nat Commun. 2020; 11:123. 10.1038/s41467-019-13981-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Larsen DH, Hari F, Clapperton JA et al. The NBS1-treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat Cell Biol. 2014; 16:792–803. 10.1038/ncb3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Korsholm LM, Gál Z, Nieto B et al. Recent advances in the nucleolar responses to DNA double-strand breaks. Nucleic Acids Res. 2020; 48:9449–61. 10.1093/nar/gkaa713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Korsholm LM, Gál Z, Lin L et al. Double-strand breaks in ribosomal RNA genes activate a distinct signaling and chromatin response to facilitate nucleolar restructuring and repair. Nucleic Acids Res. 2019; 47:8019–35. 10.1093/nar/gkz518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Velichko AK, Ovsyannikova N, Petrova NV et al. Treacle and TOPBP1 control replication stress response in the nucleolus. J Cell Biol. 2021; 220:e202008085. 10.1083/jcb.202008085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fregoso OI, Ahn J, Wang C et al. Evolutionary toggling of vpx/vpr specificity results in divergent recognition of the restriction factor SAMHD1. PLoS Pathog. 2013; 9:e1003496. 10.1371/journal.ppat.1003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berkovich E, Monnat RJ, Kastan MB Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007; 9:683–90. 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- 39. Fregoso OI, Emerman M Activation of the DNA damage response is a conserved function of HIV-1 and HIV-2 vpr that is independent of SLX4 recruitment. mBio. 2016; 7:e01433-16. 10.1128/mBio.01433-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gray SJ, Choi VW, Asokan A et al. Production of recombinant adeno-associated viral vectors and use in in vitro and in vivo administration. Curr Protoc Neurosci. 2011; Chapter 4:Unit 4.17. 10.1002/0471142301.ns0417s57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aurnhammer C, Haase M, Muether N et al. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Human Gene Therapy Methods. 2012; 23:18–28. 10.1089/hgtb.2011.034. [DOI] [PubMed] [Google Scholar]
- 42. Stringer BW, Day BW, D’Souza RCJ et al. A reference collection of patient-derived cell line and xenograft models of proneural, classical and mesenchymal glioblastoma. Sci Rep. 2019; 9:4902. 10.1038/s41598-019-41277-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dean PN, Jett JH Mathematical analysis of DNA distributions derived from flow microfluorometry. J Cell Biol. 1974; 60:523–7. 10.1083/jcb.60.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fox MH A model for the computer analysis of synchronous DNA distributions obtained by flow cytometry. Cytometry. 1980; 1:71–7. 10.1002/cyto.990010114. [DOI] [PubMed] [Google Scholar]
- 45. Potapova TA, Unruh JR, Conkright-Fincham J et al. Distinct states of nucleolar stress induced by anticancer drugs. eLife. 2023; 12:RP88799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Picard L, Ganivet Q, Allatif O et al. DGINN, an automated and highly-flexible pipeline for the detection of genetic innovations on protein-coding genes. Nucleic Acids Res. 2020; 48:e103. 10.1093/nar/gkaa680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Löytynoja A, Goldman N Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science. 2008; 320:1632–5. 10.1126/science.1158395. [DOI] [PubMed] [Google Scholar]
- 48. Kosakovsky Pond SL, Posada D, Gravenor MB et al. Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol. 2006; 23:1891–901. 10.1093/molbev/msl051. [DOI] [PubMed] [Google Scholar]
- 49. Edgar RC MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004; 32:1792–7. 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Murrell B, Weaver S, Smith MD et al. Gene-wide identification of episodic selection. Mol Biol Evol. 2015; 32:1365–71. 10.1093/molbev/msv035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang Z PAML 4: phylogenetic analysis by Maximum likelihood. Mol Biol Evol. 2007; 24:1586–91. 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 52. Guéguen L, Gaillard S, Boussau B et al. Bio++: efficient extensible libraries and tools for computational molecular evolution. Mol Biol Evol. 2013; 30:1745–50. 10.1093/molbev/mst097. [DOI] [PubMed] [Google Scholar]
- 53. Murrell B, Wertheim JO, Moola S et al. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012; 8:e1002764. 10.1371/journal.pgen.1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Murrell B, Moola S, Mabona A et al. FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol Biol Evol. 2013; 30:1196–205. 10.1093/molbev/mst030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Greenwood EJD, Williamson JC, Sienkiewicz A et al. Promiscuous targeting of cellular proteins by Vpr drives systems-level proteomic remodeling in HIV-1 infection. Cell Rep. 2019; 27:1579–96. 10.1016/j.celrep.2019.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Johnson JR, Crosby DC, Hultquist JF et al. Global post-translational modification profiling of HIV-1-infected cells reveals mechanisms of host cellular pathway remodeling. Cell Rep. 2022; 39:110690. 10.1016/j.celrep.2022.110690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Subramanian A, Tamayo P, Mootha VK et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005; 102:15545–50. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shannon P, Markiel A, Ozier O et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Szklarczyk D, Kirsch R, Koutrouli M et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023; 51:D638–46. 10.1093/nar/gkac1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen M, Elder RT, Yu M et al. Mutational analysis of Vpr-induced G2 arrest, nuclear localization, and cell death in fission yeast. J Virol. 1999; 73:3236–45. 10.1128/JVI.73.4.3236-3245.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vodicka MA, Koepp DM, Silver PA et al. HIV-1 Vpr interacts with the nuclear transport pathway to promote macrophage infection. Genes Dev. 1998; 12:175–85. 10.1101/gad.12.2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kawano K, Doucet AJ, Ueno M et al. HIV-1 vpr and p21 restrict LINE-1 mobility. Nucleic Acids Res. 2018; 46:8454–70. 10.1093/nar/gky688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Martins L, DePaula-Silva AB, Planelles V HIV-1 Vpr induces degradation of nucleolar protein CCDC137 as a consequence of cell cycle arrest. bioRxiv16 March 2021, preprint: not peer reviewed 10.1101/2021.03.16.435666. [DOI]
- 64. Dobransky A, Root M, Hafner N et al. CRL4-DCAF1 Ubiquitin ligase dependent functions of HIV viral protein R and viral protein X. Viruses. 2024; 16:1313. 10.3390/v16081313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guenzel CA, Hérate C, Benichou S HIV-1 Vpr—a still “enigmatic multitasker”. Front Microbiol. 2014; 5:127. 10.3389/fmicb.2014.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hrecka K, Hao C, Gierszewska M et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011; 474:658–61. 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Laguette N, Sobhian B, Casartelli N et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011; 474:654–7. 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tajrishi MM, Tuteja R, Tuteja N Nucleolin: the most abundant multifunctional phosphoprotein of nucleolus. Commun Integr Biol. 2011; 4:267–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Raška I, Koberna K, Malínský J et al. The nucleolus and transcription of ribosomal genes. Biol Cell. 2004; 96:579–94. 10.1016/j.biolcel.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 70. Sharp PM, Hahn BH The evolution of HIV-1 and the origin of AIDS. Phil Trans R Soc B. 2010; 365:2487–94. 10.1098/rstb.2010.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wu RS, Kumar A, Warner JR Ribosome formation is blocked by camptothecin, a reversible inhibitor of RNA synthesis. Proc Natl Acad Sci USA. 1971; 68:3009–14. 10.1073/pnas.68.12.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Han T, Tong J, Wang M et al. Olaparib induces RPL5/RPL11-dependent p53 activation via nucleolar stress. Front Oncol. 2022; 12:821366. 10.3389/fonc.2022.821366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pietrzak M, Smith SC, Geralds JT et al. Nucleolar disruption and apoptosis are distinct neuronal responses to etoposide-induced DNA damage. J Neurochem. 2011; 117:1033–46. 10.1111/j.1471-4159.2011.07279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Carotenuto P, Pecoraro A, Palma G et al. Therapeutic approaches targeting nucleolus in cancer. Cells. 2019; 8:1090. 10.3390/cells8091090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hua L, Yan D, Wan C et al. Nucleolus and nucleolar stress: from cell fate decision to disease development. Cells. 2022; 11:3017. 10.3390/cells11193017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sutton EC, DeRose VJ Early nucleolar responses differentiate mechanisms of cell death induced by oxaliplatin and cisplatin. J Biol Chem. 2021; 296:100633. 10.1016/j.jbc.2021.100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zgheib S, Taha N, Zeiger M et al. The human cellular protein NoL12 is a specific partner of the HIV-1 nucleocapsid protein NCp7. J Virol. 2023; 97:e0004023. 10.1128/jvi.00040-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Milev MP, Ravichandran M, Khan MF et al. Characterization of staufen1 ribonucleoproteins by mass spectrometry and biochemical analyses reveal the presence of diverse host proteins associated with human immunodeficiency virus type 1. Front Microbiol. 2012; 3:367. 10.3389/fmicb.2012.00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kleinman CL, Doria M, Orecchini E et al. HIV-1 infection causes a down-regulation of genes involved in ribosome biogenesis. PLoS One. 2014; 9:e113908. 10.1371/journal.pone.0113908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gadad SS, Rajan RE, Senapati P et al. HIV-1 infection induces acetylation of NPM1 that facilitates Tat localization and enhances viral transactivation. J Mol Biol. 2011; 410:997–1007. 10.1016/j.jmb.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 81. Yang K, Yang J, Yi J Nucleolar Stress: hallmarks, sensing mechanism and diseases. Cell Stress. 2018; 2:125–40. 10.15698/cst2018.06.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yan X, Kuang B-H, Ma S et al. NOP14-mediated ribosome biogenesis is required for mTORC2 activation and predicts rapamycin sensitivity. J Biol Chem. 2024; 300:105681. 10.1016/j.jbc.2024.105681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gérus M, Bonnart C, Caizergues-Ferrer M et al. Evolutionarily conserved function of RRP36 in early cleavages of the pre-rRNA and production of the 40S ribosomal subunit. Mol Cell Biol. 2010; 30:1130–44. 10.1128/MCB.00999-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Dragon F, Gallagher JEG, Compagnone-Post PA et al. A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature. 2002; 417:967–70. 10.1038/nature00769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Iapalucci-Espinoza S, Franze-Fernández MT Effect of protein synthesis inhibitors and low concentrations of actinomycin D on ribosomal RNA synthesis. FEBS Lett. 1979; 107:281–4. 10.1016/0014-5793(79)80390-7. [DOI] [PubMed] [Google Scholar]
- 86. Chédin S, Laferté A, Hoang T et al. Is ribosome synthesis controlled by pol I transcription?. Cell Cycle. 2007; 6:11–5. 10.4161/cc.6.1.3649. [DOI] [PubMed] [Google Scholar]
- 87. Nechay M, Wang D, Kleiner RE Inhibition of nucleolar transcription by oxaliplatin involves ATM/ATR kinase signaling. Cell Chem Biol. 2023; 30:906–19. 10.1016/j.chembiol.2023.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. van Sluis M, McStay B A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes Dev. 2015; 29:1151–63. 10.1101/gad.260703.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Li J, Zhao H, McMahon A et al. APE1 assembles biomolecular condensates to promote the ATR–Chk1 DNA damage response in nucleolus. Nucleic Acids Res. 2022; 50:10503–25. 10.1093/nar/gkac853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ma H, Pederson T The nucleolus stress response is coupled to an ATR-Chk1–mediated G2 arrest. MBoC. 2013; 24:1334–42. 10.1091/mbc.e12-12-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sakai K, Dimas J, Lenardo MJ The Vif and Vpr accessory proteins independently cause HIV-1-induced T cell cytopathicity and cell cycle arrest. Proc Natl Acad Sci USA. 2006; 103:3369–74. 10.1073/pnas.0509417103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Iijima K, Kobayashi J, Ishizaka Y Structural alteration of DNA induced by viral protein R of HIV-1 triggers the DNA damage response. Retrovirology. 2018; 15:8. 10.1186/s12977-018-0391-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Szaflarski W, Leśniczak-Staszak M, Sowiński M et al. Early rRNA processing is a stress-dependent regulatory event whose inhibition maintains nucleolar integrity. Nucleic Acids Res. 2022; 50:1033–51. 10.1093/nar/gkab1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Scull CE, Schneider DA Coordinated control of rRNA processing by RNA polymerase I. Trends Genet. 2019; 35:724–33. 10.1016/j.tig.2019.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zisi A, Bartek J, Lindström MS Targeting ribosome biogenesis in cancer: lessons learned and way forward. Cancers. 2022; 14:2126. 10.3390/cancers14092126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Salvetti A, Greco A Viruses and the nucleolus: the fatal attraction. Biochim Biophys Acta. 2014; 1842:840–7. 10.1016/j.bbadis.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Callé A, Ugrinova I, Epstein AL et al. Nucleolin is required for an efficient herpes simplex virus type 1 infection. J Virol. 2008; 82:4762–73. 10.1128/JVI.00077-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chen H, Wurm T, Britton P et al. Interaction of the coronavirus nucleoprotein with nucleolar antigens and the host cell. J Virol. 2002; 76:5233–50. 10.1128/JVI.76.10.5233-5250.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dove BK, You J, Reed ML et al. Changes in nucleolar morphology and proteins during infection with the coronavirus infectious bronchitis virus. Cell Microbiol. 2006; 8:1147–57. 10.1111/j.1462-5822.2006.00698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Michienzi A, De Angelis FG, Bozzoni I et al. A nucleolar localizing rev binding element inhibits HIV replication. AIDS Res Ther. 2006; 3:13. 10.1186/1742-6405-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Arizala JAC, Takahashi M, Burnett JC et al. Nucleolar localization of HIV-1 rev is required, yet insufficient for production of infectious viral particles. AIDS Res Hum Retroviruses. 2018; 34:961–81. 10.1089/aid.2017.0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Michienzi A, Li S, Zaia JA et al. A nucleolar TAR decoy inhibitor of HIV-1 replication. Proc Natl Acad Sci USA. 2002; 99:14047–52. 10.1073/pnas.212229599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ponti D, Troiano M, Bellenchi GC et al. The HIV Tat protein affects processing of ribosomal RNA precursor. BMC Cell Biol. 2008; 9:32. 10.1186/1471-2121-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Jarboui MA, Bidoia C, Woods E et al. Nucleolar protein trafficking in response to HIV-1 tat: rewiring the nucleolus. PLoS One. 2012; 7:e48702. 10.1371/journal.pone.0048702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Iarovaia OV, Ioudinkova ES, Velichko AK et al. Manipulation of cellular processes via nucleolus hijaking in the course of viral infection in mammals. Cells. 2021; 10:1597. 10.3390/cells10071597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Sharma A, Yilmaz A, Marsh K et al. Thriving under stress: selective translation of HIV-1 structural protein mRNA during vpr-mediated impairment of eIF4E translation activity. PLoS Pathog. 2012; 8:e1002612. 10.1371/journal.ppat.1002612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Jin J, Bai H, Yan H et al. PRMT2 promotes HIV-1 latency by preventing nucleolar exit and phase separation of Tat into the Super Elongation Complex. Nat Commun. 2023; 14:7274. 10.1038/s41467-023-43060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The CCDC137 alignment data underlying this article are available in FigShare at https://figshare.com/ and can be accessed with https://doi.org/10.6084/m9.figshare.26841640 and https://doi.org/10.6084/m9.figshare.26841634.