










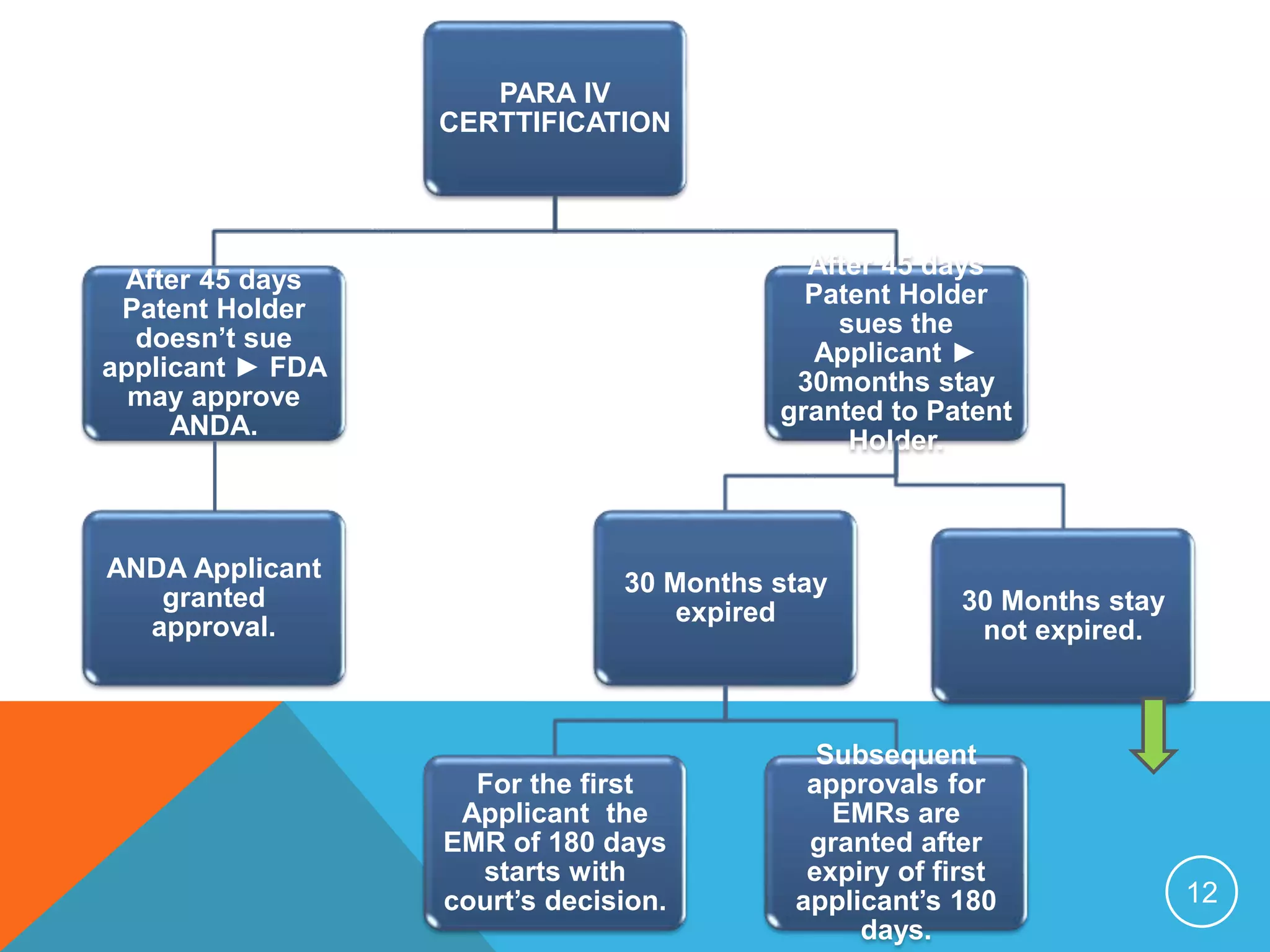





























The document discusses generic drugs and the Abbreviated New Drug Application (ANDA) process. It explains that an ANDA contains data to review and approve a generic drug as a safe and effective alternative to the original brand name drug. It provides details on the Hatch-Waxman Act which established the modern generic drug approval pathway and allows generics to reference clinical data from the branded version. The document also outlines the requirements for generic drugs to be approved as well as the various certification pathways in the ANDA process.






















![Abbreviated New Drug Application [ANDA]](https://cdn.slidesharecdn.com/ss_thumbnails/abbreviatednewdrugapplicationanda-160619062810-thumbnail.jpg?width=640&height=640&fit=bounds)












































