BITS: Basics of sequence analysis
10 likes•9,183 views

This document outlines various concepts in sequence analysis within bioinformatics. It discusses tools and methodologies for analyzing DNA, RNA, and protein sequences, including prediction of biological functions, metrics for sequence properties, and the performance evaluation of prediction tools. Additionally, it emphasizes the importance of using reliable databases and tools for automatic annotation and feature prediction, alongside offering insights into specific analysis techniques like sequence alignment and motif detection.
















![Regular expressions / patterns are the
simplest way to represent motifs
A representation of all residues with equal probability.
123456 Position: 1. 2. 3. 4. 5. 6.
ATPKAE
KKPKAA [AKT] [AKLT] P [AK] [APT] [ADEKT-]
AKPKAK
TKPKPA
AKPKT-
AKPAAK ? Does this sequence match: AKPKTE
KLPKAD V V V V V V
AKPKAA
Consensus: AKPKAA ? And this sequence: KKPETE
V V V X V V
? And what about this one: TLPATE
For every position the most V V V V V V
Frequently occurring residue](https://image.slidesharecdn.com/mod3-introh12011seqan-110627023719-phpapp01/85/BITS-Basics-of-sequence-analysis-18-320.jpg)







-H-S-{P}-x(2)-P-x(2,4)-C>
You can retrieve sequences which correspond to a pattern,
you made up yourself, observed in an alignment or an
known one. The syntax is specific, but not difficult: see
link below!
http://prosite.expasy.org/scanprosite/scanprosite-doc.html#pattern_syntax](https://image.slidesharecdn.com/mod3-introh12011seqan-110627023719-phpapp01/85/BITS-Basics-of-sequence-analysis-26-320.jpg)

















![Sequence logos can give an insight in
the important residues of binding sites
DNA: an entry from JASPAR: tata box
A [ 61 16 352 3 354 268 360 222 155 56 83 82 82 68 77 ]
C [145 46 0 10 0 0 3 2 44 135 147 127 118 107 101 ]
G [152 18 2 2 5 0 10 44 157 150 128 128 128 139 140 ]
T [ 31 309 35 374 30 121 6 121 33 48 31 52 61 75 71 ]](https://image.slidesharecdn.com/mod3-introh12011seqan-110627023719-phpapp01/85/BITS-Basics-of-sequence-analysis-46-320.jpg)









BITS: Basics of sequence analysis
- 1. Basic bioinformatics concepts, databases and tools Module 3 Sequence analysis Joachim Jacob http://www.bits.vib.be Updated Feb 2012 http://dl.dropbox.com/u/18352887/BITS_training_material/Link%20to%20mod3-intro_H1_2012_SeqAn.pdf
- 2. In this third module, we will discuss the possible analyses of sequences Module 1 Sequence databases and keyword searching Module 2 Sequence similarity Module 3 Sequence analysis: types, interpretation, results
- 3. In this third module, we will discuss the possible analyses of sequences
- 4. Sequence analysis tries to read sequences to infer biological properties AGCTACTACGGACTACTAGCAGCTACCTCTCTG - is this coding sequence? - can this sequence bind a certain TF? - what is the melting temperature? - what is the GC content? - does it fold into a stable secondary structure? …
- 5. Tools that can predict a biological feature are trained with examples Automatic annotation vs. experimentally verified annotations - Training dataset of sequences (← exp. verified) - An algorithm defines parameters used for prediction - The algorithm determines/classifies whether the sequence(s) contains the feature (→ automatic annotation)
- 6. The assumption to being able to read biological function is the central paradigm DNA → protein sequence → structure → activity (binding, enzymatic activity, regulatory,...) So the premise to do analysis: biological function can be read from the (DNA) sequence. Predictions always serve as a basis for further experiments.
- 7. Analysis can be as simple as measuring properties or predicting features Protein − Metrics (e.g. how many alanines in my seq) − Modifications and other predictions − Domains and motifs DNA/RNA − Metrics (e.g. how many GC) − Predicting Gene prediction Promotor Structure
- 8. Simple protein sequence analysis One might be interested in: pI (isoelectric point) prediction Composition metrics Hydrophobicity calculation Reverse translation (protein → dna) Occurrence of simple patterns (e.g. does KDL occurs and how many times) ... http://en.wikipedia.org/wiki/Hydrophobicity_scales http://www.sigmaaldrich.com/life-science/metabolomics/learning-center/amino-acid-reference-cha
- 9. Protein sequence analysis tools are gathered on Expasy http://www.expasy.org/tools (SIB) Others: http://www.ebi.ac.uk/Tools/protein.html http://bioweb.pasteur.fr/protein/intro-en.html SMS2
- 10. Never trust a tool's output blindly Interpreting depends on the kind of output When a prediction result is obtained, the question arises 'Is it true?' (in biological sense) Programs giving a 'binary' Programs giving score/P- result: 1 or 0, a hit or a miss. value result: the chance that the 'result' is 'not real' → the Approach: You should lower, the better comparing different prediction programs for Approach: asses the p-value higher confidence. E.g. ScanProsite for a motif E.g. SignalP for signal peptide prediction.
- 11. The basis for the prediction of features is nearly always a sequence alignment Based on experimentally verified sequence annotations, a multiple sequence alignment is constructed Different methods exist to capture the information gained from this multiple sequence alignment
- 12. Alignment reveals similar residues which can indicate identical structure Same structure, hence most likely same function Most protein pairs with more than 25- 30 out of 100 identical residues were Chances are that found to be structurally similar. the structure is not Also proteins with <10% identity can the same have similar structure. http://peds.oxfordjournals.org/content/12/2/85.long
- 13. The structure of a protein sequence determines his biological function Number of Primary = AA chain Reported structures Feb 2012: ~ 535 000 in Swissprot Secondary = structural entities (helix, beta-strands, beta-sheets, loops) Tertiary = 3D Nov 2011: ~ 80 000 in PDB Quaternary = interactions http://en.wikipedia.org/wiki/Protein_structure
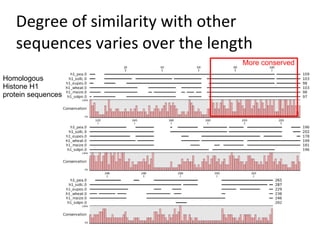
- 14. Degree of similarity with other sequences varies over the length More conserved Homologous Histone H1 protein sequences
- 15. Protein sequences can consist of structurally different parts Domain part of the tertiary structure of a protein that can exist, function and evolve independently of the rest, linked to a certain biological function Motif part (not necessarily contiguous) of the primary structure of a protein that corresponds to the signature of a biological function. Can be associated with a domain. Feature part of the sequence for which some annotation has been added. Some features correspond to domain or motif assignments.
- 16. Based on motifs and domains, proteins are assigned to families Nearly synonymous with gene family Evolutionary related proteins Significant structural similarity of domains is reflected in sequence similarity, and is due to a common ancestral sequence part, resulting in domain families.
- 17. Domains and motifs are represented by simple and complex methods domain Gapped alignment Motif/domain in silico can be represented by 1. Regular expression / pattern 2. Frequency matrix / profile 3. Machine learning techniques : Hidden Markov Model http://bioinfo.uncc.edu/zhx/binf8312/lecture-7-SequenceAnalyses.pdf
- 18. Regular expressions / patterns are the simplest way to represent motifs A representation of all residues with equal probability. 123456 Position: 1. 2. 3. 4. 5. 6. ATPKAE KKPKAA [AKT] [AKLT] P [AK] [APT] [ADEKT-] AKPKAK TKPKPA AKPKT- AKPAAK ? Does this sequence match: AKPKTE KLPKAD V V V V V V AKPKAA Consensus: AKPKAA ? And this sequence: KKPETE V V V X V V ? And what about this one: TLPATE For every position the most V V V V V V Frequently occurring residue
- 19. Frequency matrices or profiles include the chance of observing the residues For every position of a motif, a list of all amino acids is made with their frequency. Position-specific weight/scoring matrix or profile. More sensitive way. Profile 123456 Position: 1. 2. 3. 4. 5. 6. ATPKAE KKPKAA A 0.625 0 0 1/8 6/8 3/8 AKPKAK D 0 0 0 0 0 1/8 TKPKPA E 0 0 0 0 0 1/8 AKPKT- K 0.25 6/8 0 7/8 0 2/8 AKPAAK L 0 1/8 0 0 0 0 KLPKAD P 0 0 1 0 1/8 0 AKPKAA T 1/8 1/8 0 0 1/8 0 Consensus: AKPKA- - 0 0 0 0 0 1/8 Sum 1 1 1 1 1 1 ? Query: AKPKTE ? Query: KKPETE ? Query: TLPATE Example: http://expasy.org/prosite/PS51092 http://prosite.expasy.org/prosuser.html#meth2
- 20. How good a sequence matches a profile is reported with a score PSWM: scores 123456 Position: 1. 2. 3. 4. 5. 6. ATPKAE KKPKAA A 2.377 -2.358 -2.358 0.257 2.631 1.676 AKPKAK D -2.358 -2.358 -2.358 -2.358 -2.358 0.257 TKPKPA E -2.358 -2.358 -2.358 -2.358 -2.358 0.257 AKPKT- K 1.134 2.631 -2.358 2.847 -2.358 1.134 AKPAAK L -2.358 0.257 -2.358 -2.358 -2.358 -2.358 P -2.358 -2.358 0.257 -2.358 0.257 -2.358 KLPKAD T 0.257 0.257 -2.358 -2.358 0.257 -2.358 AKPKAA Consensus: AKPKA- ? Query: AKPKTE Score = 11.4 ? Query: KKPETE Score = 5.0 ? Query: TLPATE Score = 4.3 http://prosite.expasy.org/prosuser.html#meth2
- 21. A hidden Markov Model takes also into account the gaps in an alignment The schematic representation of a HMM http://www.myoops.org/twocw/mit/NR/rdonlyres/Electrical-Engineering-
- 22. Building a HMM from a multiple sequence alignment
- 23. Use HMMER to very sensitively search protein database with a HMM You can search with a profile in a sequence database
- 24. Some profile adjustments to the BLAST protocol exist for particular purposes PSI-BLAST to identify distantly related proteins PSI-BLAST (position specific iterated) After a search result, a profile is made of the similar sequences, and this is used again to search a database PHI-BLAST protein with matching of a pattern PHI-BLAST (pattern hit initiated): you provide a pattern, which all BLAST results should satisfy. CSI-BLAST is more sensitive than PSI-BLAST in identifying distantly related proteins PSI BLAST http://www.ncbi.nlm.nih.gov/blast/Blast.cgi?CMD=Web&PAGE=Proteins&PROGRAM=b PHI BLAST http://www.ncbi.nlm.nih.gov/blast/Blast.cgi?CMD=Web&PAGE=Proteins&PROGRAM=b CSI BLAST http://toolkit.tuebingen.mpg.de/cs_blast
- 25. Many databases exist that keep patterns, profiles or models related to function Motif / domain databases (see NCBI bookshelf for good overview) http://www.ebi.ac.uk/interpro/ - integrated db http://expasy.org/prosite/ (motifs) PFAM – hidden markov profiles (domains) CDD (Conserved domains database) (NCBI - integrated) Prodom (domain) (automatic extraction) SMART (domain) PRINTS (motif) sets of local alignments without gaps, used as frequency matrices, made by searching manually made "seed alignments" against UniProt sequences
- 26. Prosite is a database gathering patterns from sequence alignments ScanProsite tool : search the prosite database for a pattern ( present or not ) Example : [DE](2)-H-S-{P}-x(2)-P-x(2,4)-C> You can retrieve sequences which correspond to a pattern, you made up yourself, observed in an alignment or an known one. The syntax is specific, but not difficult: see link below! http://prosite.expasy.org/scanprosite/scanprosite-doc.html#pattern_syntax
- 27. Interpro classifies the protein data into families based on the domain and motifs Interpro takes all existing motif and domains databases as input ('signatures'), and aligns them to create protein domain families. This reduces redundancy. Each domain is than given an identifier IPRxxxxxxx. Uneven size of motifs and families between families are handled by 'relations' : parent - child and contains - found in Families,... Regions, domains, ... http://www.ebi.ac.uk/interpro/user_manual.html#type
- 28. Interpro summarizes domains and motifs from a dozen of domain databases http://www.ebi.ac.uk/interpro/databases.html ftp://ftp.ebi.ac.uk/pub/software/unix/iprscan/README.html#2
- 29. InterPro entries are grouped in types Family Entries span complete sequence Domain Biologically functional units Repeat Region Conserved site Active site Binding site PTM site
- 30. InterPro entries are grouped in types
- 31. You can search your sequence for known domains on InterProScan Interproscan http://www.ebi.ac.uk/Tools/pfa/iprscan/
- 32. A sequence logo provides a visual summary of a motif Creating a sequence logo Create a nicely looking logo of a motif sequence: size of letters indicated frequency. Weblogo - a basic web application to create colorful logo's IceLogo - a powerful web application to create customized logo's
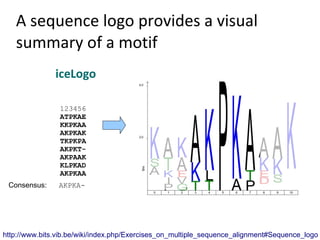
- 33. A sequence logo provides a visual summary of a motif iceLogo 123456 ATPKAE KKPKAA AKPKAK TKPKPA AKPKT- AKPAAK KLPKAD AKPKAA Consensus: AKPKA- http://www.bits.vib.be/wiki/index.php/Exercises_on_multiple_sequence_alignment#Sequence_logo
- 34. There is always a chance that a prediction of a feature by a tool is false Number Number of of matches matches True negatives True negatives True positives True positives Score Score Threshold Threshold False negatives False positives Ideal situation Reality of the databases
- 35. Assessing the performance of categorizing tools with sensitivity and specificity PREDICTION “Confusion matrix” Feature is Feature is predicted NOT predicted True False Sequence contains feature positive Negatives “Type I error” TRUTH False True Sequence does NOT contain feature positive negative “Type II error”
- 36. Assessing the performance of categorizing tools with sensitivity and specificity PREDICTION “Confusion matrix” Feature is Feature is predicted NOT predicted Sensitivity Sequence contains feature True positives/(TP + FN) TRUTH False True Sequence does NOT contain feature positive negative
- 37. Assessing the performance of categorizing tools with sensitivity and specificity PREDICTION “Confusion matrix” Feature is Feature is predicted NOT predicted Sequence contains feature TRUTH Selectivity Sequence does NOT contain feature or Specificity TN/(FP + TN)
- 38. Assessing the performance of categorizing tools with sensitivity and specificity PREDICTION “Confusion matrix” Feature is Feature is predicted NOT predicted Sequence contains feature error rate* TRUTH FP+FN/total Sequence does NOT contain feature * misclassification rate
- 39. Assessing the performance of categorizing tools with sensitivity and specificity PREDICTION “Confusion matrix” Feature is Feature is predicted NOT predicted Sequence contains feature Accuracy TRUTH TP+TN/total Sequence does NOT contain feature
- 40. Protein sequences can be searched for potential modifications http://www.expasy.org/tools/ e.g. modification (phosphorylation, acetylation,...) To deal with the confidence in the results, try different tools, and make a graph (venn diagram) to compare the results E.g. predict secreted proteins by signalP and RPSP, combine results in Venn − http://bioinformatics.psb.ugent.be/webtools/Venn/ − http://www.cmbi.ru.nl/cdd/biovenn/ Overview SignalPeptide prediction tools: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2788353/
- 41. Protein sequences can be searched for secondary structural elements Based on know structures, machine learning models of secondary structure elements are made and can be searched for. See http://bioinf.cs.ucl.ac.uk/psipred/
- 42. In case of multiple analyses on multiple sequences, mark instead of filter Starting set of sequences Worse Better Analysis filter 1 Analysis filter 1 Analysis filter 2 Analysis filter 3 Analysis filter 2 Analysis filter 3 After performing all analyses on all sequences, different filters on the results can be applied (e.g. secreted sequence, ! phosphorylated and containing a motif)
- 43. NA sequences
- 44. NA sequence analyses GC% http://mobyle.pasteur.fr/cgi-bin/portal.py?#forms::geecee Melting temperature For primer development, such as with Primer3 Structure Codon usage Codon usage table with cusp Codon adaptation index calculation with cai ... A lot of tools can be found at the Mobyle Portal:
- 45. Profiles and models are being used to model biological function in NA seqs To detect Transcription factor binding sites TRANSFAC : commercial (BIOBASE, Wolfenbüttel, Germany), started as work of Edgard Wingender, contains eukaryotic binding sites as consensus sequences and as PSSMs. Also TRANSCompel with modules of binding sites. ooTFD : commercial (IFTI, Pittsburgh PA, USA), started as work of David Gosh, contains prokaryotic and eukaroytic binding sites as consensus sequences and as PSSMs. JASPAR : open access, only representative sets of higher eukaryote binding sites as PSSMs. Can be searched against sequence or sequence pair at Consite. OregAnno : open access, collection of individual eukaryotic binding sites with their localization in the genome PAZAR : collection of open access TF databanks
- 46. Sequence logos can give an insight in the important residues of binding sites DNA: an entry from JASPAR: tata box A [ 61 16 352 3 354 268 360 222 155 56 83 82 82 68 77 ] C [145 46 0 10 0 0 3 2 44 135 147 127 118 107 101 ] G [152 18 2 2 5 0 10 44 157 150 128 128 128 139 140 ] T [ 31 309 35 374 30 121 6 121 33 48 31 52 61 75 71 ]
- 47. The RNA world has the Vienna servers http://rna.tbi.univie.ac.at/ − secondary structure prediction of ribosomal sequences − siRNA design
- 48. RNA families can be modeled by conserved bases and structure RNA motifs (http://rfam.sanger.ac.uk/search) Rfam is a databank of RNA motifs and families. It is made at the Sanger Centre (Hinxton, UK), from a subset of EMBL (well-annotated standard sequences excluding synthetic sequences + the WGS) using the INFERNAL suite of Soan Eddy. It contains local alignments with gaps with included secondary structure annotation + CMs.
- 49. Some interesting links Nucleic acid structure Unafold - Program accessible through webinterface After designing primers, you might want to check whether the primer product does (not) adapt a stable secondary structure. Some collections of links − Good overview at http://www.imb-jena.de/RNA.html − European Ribosomale RNA database (VIB PSB)
- 50. Prediction of genes in genomes rely on the integration of multiple signals Signals surrounding the gene (transcription factor binding sites, promoters, transcription terminators, splice sites, polyA sites, ribosome binding sites,...) → profile matching Differences in composition between coding and noncoding DNA (codon preference), the presence of an Open Reading Frame (ORF) → compositional analyses Similarity with known genes, aligning ESTs and (in translation) similarity with known proteins and the presence of protein motifs → similarity searches
- 51. Prediction of genes in genomes rely on the integration of multiple signals Signals Composition Similarity e.g. potential methylation sites (profiles) Alignment of ESTs GC
- 52. Software for prediction genes EMBOSS − simple software under EMBOSS : syco (codon frequency), wobble (%GC 3rd base), tcode (Ficket statistic : correlation between bases at distance 3) Examples of software using HMM model of gene : Wise2 : using also similarity with known proteins http://www.ebi.ac.uk/Tools/Wise2 GENSCAN : commercial (Chris Burge, Stanford U.) but free for academics, has models for human/A. thaliana/maize, used at EBI and NCBI for genome annotation http://mobyle.pasteur.fr/cgi-bin/portal.py?#forms::genscan GeneMark : commercial (GeneProbe, Atlanta GA, USA) but free for academic users, developed by Mark Borodovsky, has models for many prokaryotic and eukaryotic organisms http://exon.gatech.edu Tutorial on gene prediction http://www.embl.de/~seqanal/courses/spring00/GenePred.00.html
- 53. Short addendum about downloading files FTP, e.g. ftp://ftp.ebi.ac.uk/pub/databases/interpro/ – 'file transfer protocol' – Most browsers have integrated ftp 'client' – Free, easy to download files, possibility to resume after fails HTTP, e.g. http://www.ncbi.nlm.nih.gov/entrez Standard protocol for internet traffic, Slowest method Aspera – for large datasets (>10GB) downloads In use in the short read archive (SRA) Fastest method available currently
- 54. Conclusion Prediction vs. experimental verified Different algorithms need to be compared Predictions need to be validated by independent method Software <-> Databases Questions? Get social! → www.seqanswers.com → http://biostar.stackexchange.com Always only basis for further wet-lab research
- 55. Summary In this third module, we will discuss the possible analyses of sequences Sequence analysis tries to read sequences to infer biological properties Tools that can predict a biological feature are trained with examples The assumption to being able to read biological function is the central paradigm Analysis can be as simple as measuring properties or predicting features Protein sequence analysis tools are gathered on Expasy Never trust a tool's output blindly The basis for the prediction of features is nearly always a sequence alignment Alignment reveals similar residues which can indicate identical structure The structure of a protein sequence determines his biological function Degree of similarity with other sequences varies over the length Protein sequences can consist of structurally different parts Based on motifs and domains, proteins are assigned to families Domains and motifs are represented by simple and complex methods Regular expressions / patterns are the simplest way to represent motifs Frequency matrices or profiles include the chance of observing the residues How good a sequence matches a profile is reported with a score A hidden Markov Model takes also into account the gaps in an alignment Use HMMER to very sensitively search protein database with a HMM Some profile adjustments to the BLAST protocol exist for particular purposes Many databases exist that keep patterns, profiles or models related to function Prosite is a database gathering patterns from sequence alignments Interpro classifies the protein data into families based on the domain and motifs Interpro summarizes domains and motifs from a dozen of domain databases You can search your sequence for known domains on InterProScan A sequence logo can provide a visual summary of a motif Protein sequences can be searched for potential modifications Protein sequences can be searched for secondary structural elements In case of multiple analyses on multiple sequences, mark instead of filter Profiles and models are being used to model biological function in NA seqs Sequence logos can give an insight in the important residues of binding sites The RNA world has the Vienna servers RNA families can be modeled by conserved bases and structure Prediction of genes in genomes rely on the integration of multiple signals